Exhibit 99.1
TABLE OF CONTENTS
BASIS OF PRESENTATION
Unless otherwise stated, the information in this AIF is as of March 15, 2019.
In this AIF, unless stated otherwise or the context requires, all references to “$ or “US$” are to the lawful currency of the United States and all references to “CDN$” are to the lawful currency of Canada.
On March 15, 2019 the exchange rate for conversion of US dollars into Canadian dollars was US$1.00 = CDN$1.3342 based upon the Bank of Canada closing rate.
Market data and certain industry forecasts used in this AIF were obtained from market research, publicly available information and industry publications. We believe that these sources are generally reliable, but the accuracy and completeness of this information is not guaranteed. We have not independently verified such information, and we do not make any representation as to the accuracy of such information.
In this AIF, unless the context otherwise requires, references to “we”, “us”, “our” or similar terms, as well as references to “Aurinia” or the “Company”, refer to Aurinia Pharmaceuticals Inc., together with our subsidiaries.
This AIF describes the Company and its operations, its prospects, risks and other factors that affect its business.
Capitalized terms that are not otherwise defined in this AIF have the meanings attributed thereto in Schedule 3 to this AIF.
FORWARD-LOOKING STATEMENTS
A statement is forward-looking when it uses what we know and expect today to make a statement about the future. Forward-looking statements may include words such as “anticipate”, “believe”, “intend”, “expect”, “goal”, “may”, “outlook”, “plan”, “seek”, “project”, “should”, “strive”, “target”, “could”, “continue”, “potential” and “estimated”, or the negative of such terms or comparable terminology. You should not place undue reliance on the forward-looking statements, particularly those concerning anticipated events relating to the development, clinical trials, regulatory approval, and marketing of our products and the timing or magnitude of those events, as they are inherently risky and uncertain.
Securities laws encourage companies to disclose forward-looking information so that investors can get a better understanding of our future prospects and make informed investment decisions. These statements made in this AIF may include, without limitation:
| |
• | our belief that the AURA clinical trial had positive results; |
| |
• | our belief that we have sufficient cash resources to adequately fund operations; |
| |
• | our belief that the totality of data from both the AURORA and AURA clinical trials can potentially serve as the basis for a NDA submission with the FDA following a successful completion of the AURORA clinical trial; |
| |
• | our belief that confirmatory data generated from the single AURORA clinical trial and the completed AURA clinical trial should support regulatory submissions in the United States, Europe and Japan and the timing of such including the NDA submission in the United States; |
| |
• | our belief that granted formulation patents regarding the delivery of voclosporin to the ocular surface for conditions such as dry eye have the potential to be of therapeutic value; |
| |
• | our belief in the duration of patent exclusivity for voclosporin and that the patents owned by us are valid; |
| |
• | our belief in receiving extensions to patent life based on certain events or classifications; |
| |
• | our plans and expectations and the timing of commencement, enrollment, completion and release of results of clinical trials; |
| |
• | our current forecast for the cost of the AURORA clinical trial and the AURORA 2 extension trial; |
| |
• | our intention to demonstrate that voclosporin possesses pharmacologic properties with the potential to demonstrate best-in-class differentiation with first-in-class status for the treatment of LN outside of Japan; |
| |
• | our belief of the key potential benefits of voclosporin in the treatment of LN and other podocytopathies; |
| |
• | our target launch date for voclosporin as a treatment for LN in early 2021; |
| |
• | our belief in voclosporin being potentially a best-in-class CNI with robust intellectual property exclusivity and the benefits over existing commercially available CNIs; |
| |
• | our belief that CNI's are a mainstay of treatment or DES; |
| |
• | our belief that voclosporin has further potential to be effectively used across a range of therapeutic autoimmune areas including DES and FSGS; |
| |
• | the timing for completion of enrollment and for data availability for our Phase 2 clinical study for voclosporin in FSGS patients; |
| |
• | statements concerning the anticipated commercial potential of voclosporin for the treatment of LN, DES and FSGS; |
| |
• | our plan to expand voclosporin renal franchise to include FSGS; |
| |
• | our belief that the expansion of the renal franchise could create significant value for shareholders; |
| |
• | our intention to use the net proceeds from financings for various purposes; |
| |
• | our belief that our current financial resources are sufficient to fund our existing LN program including the AURORA trial and the NDA submission to the FDA, conduct the current Phase 2 study for FSGS, commence additional studies for DES, and fund operations into mid-2020; |
| |
• | our plans to generate future revenues from products licensed to pharmaceutical and biotechnology companies; |
| |
• | statements concerning partnership activities and health regulatory discussions; |
| |
• | statements concerning the potential market for voclosporin; |
| |
• | our ability to take advantage of financing opportunities if and when needed; |
| |
• | our belief that VOS has the potential to compete in the multi-billion-dollar human prescription dry eye market; |
| |
• | our intention to seek additional corporate alliances and collaborative agreements to support the commercialization and development of our products; |
| |
• | our belief that the USPTO will issue a new patent covering the dosing protocol for voclosporin in LN, with a patent term extending to 2037; |
| |
• | our belief that additional patents may be granted worldwide based on our filings under the Patent Cooperation Treaty; |
| |
• | our strategy to become a global biopharmaceutical company; |
| |
• | our plan to conduct a confirmatory drug-drug interaction study; |
| |
• | our plan to conduct a study with pediatric patients; and |
| |
• | our belief that the annualized pricing for voclosporin for LN could range between US$45,000 and US$100,000. |
Such statements reflect our current views with respect to future events and are subject to risks and uncertainties and are necessarily based on a number of estimates and assumptions that, while considered reasonable by management, as at the date of such statements, are inherently subject to significant business, economic, competitive, political, regulatory, legal, scientific and social uncertainties and contingencies, many of which, with respect to future events, are subject to change. The factors and assumptions used by management to develop such forward-looking statements include, but are not limited to:
| |
• | the assumption that we will be able to obtain approval from regulatory agencies on executable development programs with parameters that are satisfactory to us; |
| |
• | the assumption that recruitment to clinical trials will occur as projected; |
| |
• | the assumption that we will successfully complete our clinical programs on a timely basis, including conducting the required AURORA clinical trial and meet regulatory requirements for approval of marketing authorization applications and new drug approvals, as well as favourable product labeling; |
| |
• | the assumption that the planned studies will achieve positive results; |
| |
• | the assumptions regarding the costs and expenses associated with our clinical trials; |
| |
• | the assumption that regulatory requirements and commitments will be maintained; |
| |
• | the assumption that we will be able to meet GMP standards and manufacture and secure a sufficient supply of voclosporin on a timely basis to successfully complete the development and commercialization of voclosporin; |
| |
• | the assumptions on the market value for the LN program; |
| |
• | the assumption that our patent portfolio is sufficient and valid; |
| |
• | the assumption that the USPTO will issue a new patent for its dosing protocol once applicable steps have been followed and fees paid; |
| |
• | the assumption that we will be able to extend our patents to the fullest extent allowed by law, on terms most beneficial to us; |
| |
• | the assumptions on the market; |
| |
• | the assumption that there is a potential commercial value for other indications for voclosporin; |
| |
• | the assumption that market data and reports reviewed by us are accurate; |
| |
• | the assumption that another company will not create a substantial competitive product for Aurinia’s LN business without violating Aurinia’s intellectual property rights; |
| |
• | the assumptions on the burn rate of Aurinia’s cash for operations; |
| |
• | the assumption that our current good relationships with our suppliers, service providers and other third parties will be maintained; |
| |
• | the assumption that we will be able to attract and retain a sufficient amount of skilled staff; and/or |
| |
• | the assumptions relating to the capital required to fund operations through AURORA clinical trial results and regulatory submission. |
It is important to know that:
| |
• | actual results could be materially different from what we expect if known or unknown risks affect our business, or if our estimates or assumptions turn out to be inaccurate. As a result, we cannot guarantee that any forward-looking statement will materialize and, accordingly, you are cautioned not to place undue reliance on these forward-looking statements; |
| |
• | forward-looking statements do not take into account the effect that transactions or non-recurring or other special items announced or occurring after the statements are made may have on our business. For example, they do not include the effect of mergers, acquisitions, other business combinations or transactions, dispositions, sales of assets, asset write-downs or other charges announced or occurring after the forward-looking statements are made. The financial impact of such transactions and non-recurring and other special items can be complex and necessarily depend on the facts particular to each of them. Accordingly, the expected impact cannot be meaningfully described in the abstract or presented in the same manner as known risks affecting our business. |
The factors discussed below and other considerations discussed in the “Risk Factors” section of this AIF could cause our actual results to differ significantly from those contained in any forward-looking statements.
Such forward-looking statements involve known and unknown risks, uncertainties, and other factors that may cause our actual results, performance, or achievements to differ materially from any assumptions, further results, performance or achievements expressed or implied by such forward-looking statements. Important factors that could cause such differences include, among other things, the following:
| |
• | the need for additional capital in the future to continue to fund our development programs and commercialization activities, and the effect of capital market conditions and other factors on capital availability; |
| |
• | difficulties, delays, or failures we may experience in the conduct of and reporting of results of our clinical trials for voclosporin; |
| |
• | difficulties in meeting GMP standards and the manufacturing and securing of a sufficient supply of voclosporin on a timely basis to successfully complete the development and commercialization of voclosporin; |
| |
• | difficulties, delays or failures in obtaining regulatory approvals for the initiation of clinical trials; |
| |
• | difficulties in gaining alignment among the key regulatory jurisdictions, EMA, FDA and PMDA, which may require further clinical activities; |
| |
• | difficulties, delays or failures in obtaining regulatory approvals to market voclosporin; |
| |
• | not being able to extend our patent portfolio for voclosporin; |
| |
• | our patent portfolio not covering all of our proposed uses of voclosporin; |
| |
• | the uncertainty that the FDA will approve the use of voclosporin for LN and that the label for such use will follow the dosing protocol pursuant to the Notice of Allowance; |
| |
• | difficulties we may experience in completing the development and commercialization of voclosporin; |
| |
• | the market for the LN business may not be as we have estimated; |
| |
• | insufficient acceptance of and demand for voclosporin; |
| |
• | difficulties obtaining adequate reimbursements from third party payors; |
| |
• | difficulties obtaining formulary acceptance; |
| |
• | competitors may arise with similar products; |
| |
• | product liability, patent infringement and other civil litigation; |
| |
• | injunctions, court orders, regulatory and other enforcement actions; |
| |
• | we may have to pay unanticipated expenses, and/or estimated costs for clinical trials or operations may be underestimated, resulting in our having to make additional expenditures to achieve our current goals; |
| |
• | difficulties, restrictions, delays, or failures in obtaining appropriate reimbursement from payors for voclosporin; and/or |
| |
• | difficulties we may experience in identifying and successfully securing appropriate vendors to support the development and commercialization of our product. |
Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance or achievements. These forward-looking statements are made as of the date hereof.
For additional information on risks and uncertainties in respect of the Company and its business, please see the “Risks Factors” section of this AIF. Although we believe that the expectations reflected in such forward-looking statements and information are reasonable, undue reliance should not be placed on forward-looking statements or information because we can give no assurance that such expectations will prove to be correct.
OVERVIEW
Corporate structure
Aurinia is a late clinical stage biopharmaceutical company focused on developing and commercializing therapies to treat targeted patient populations that are suffering from serious diseases with a high unmet medical need. We are currently developing voclosporin, an investigational drug, for the potential treatment of LN, DES and FSGS.
Our head office is located at #1203-4464 Markham Street, Victoria, British Columbia V8Z 7X8. Aurinia has its registered office located at #201, 17904-105 Avenue, Edmonton, Alberta T5S 2H5 where the finance function is performed.
Aurinia Pharmaceuticals Inc. is organized under the Business Corporations Act (Alberta). Our Common Shares are currently listed and traded on the NASDAQ under the symbol "AUPH" and on the TSX under the symbol "AUP".
We have two wholly-owned subsidiaries: Aurinia Pharma U.S., Inc., (Delaware incorporated) and Aurinia Pharma Limited (United Kingdom incorporated).
Our By-Law No. 2 was amended at a shareholder’s meeting held on August 15, 2013 to include provisions requiring advance notice for any nominations of directors by shareholders, which are described further in our most recent information circular.
BUSINESS OF THE COMPANY
We are currently developing voclosporin, an investigational drug, for the potential treatment of LN, DES and FSGS. Voclosporin is a next generation CNI which has clinical data in over 2,400 patients across multiple indications. It has also been previously studied in kidney rejection following transplantation, psoriasis and in various forms of uveitis (an ophthalmic disease).
The topical formulation of voclosporin, VOS, is an aqueous, preservative free nanomicellar solution intended for use in the treatment of DES. Studies have been completed in rabbit and dog models. A single Phase 1 study and a Phase 2a head-to head study have also been completed in healthy volunteers and patients with DES. VOS has IP protection until 2031.
Legacy CNIs have demonstrated efficacy for a number of conditions, including transplant, DES and other autoimmune diseases; however, side effects exist which can limit their long-term use and tolerability. Some clinical complications of legacy CNIs include hypertension, hyperlipidemia, diabetes, and both acute and chronic nephrotoxicity.
Voclosporin is an immunosuppressant, with a synergistic and dual mechanism of action that has the potential to improve near- and long-term outcomes in LN when added to MMF, although not approved for such, the current standard of care for LN. By inhibiting calcineurin, voclosporin reduces cytokine activation and blocks interleukin IL-2 expression and T-cell mediated immune responses. Voclosporin also potentially stabilizes disease modifying podocytes, which protects against proteinuria. Voclosporin is made by a modification of a single amino acid of the cyclosporine molecule. This modification may result in a more predictable pharmacokinetic and pharmacodynamic relationship, an increase in potency, an altered metabolic profile, and easier dosing without the need for therapeutic drug monitoring. Clinical doses of voclosporin studied to date range from 13 - 70 mg BID. The mechanism of action of voclosporin has been validated with certain first generation CNIs for the prevention of rejection in patients undergoing solid organ transplants and in several autoimmune indications, including dermatitis, keratoconjunctivitis sicca, psoriasis, rheumatoid arthritis, and for LN in Japan.
We believe that voclosporin possesses pharmacologic properties with the potential to demonstrate best-in-class differentiation with first-in-class regulatory approval status for the treatment of LN outside of Japan.
Based on published data, we believe the key potential benefits of voclosporin in the treatment of LN and other podocytopathies are as follows:
| |
• | increased potency compared to CsA, allowing lower dosing requirements and fewer off target effects; |
| |
• | limited inter and intra patient variability, allowing for easier dosing without the need for therapeutic drug monitoring; |
| |
• | less cholesterolemia and triglyceridemia than CsA; and |
| |
• | limited incidence of glucose intolerance and diabetes at therapeutic doses compared to tacrolimus. |
Our target launch date for voclosporin as a treatment for LN is early 2021.
STRATEGY
Our business strategy is to optimize the clinical and commercial value of voclosporin and become a global biopharma company with a focused renal and autoimmune franchise. This includes the expansion of a potential renal franchise with additional renal indications and the exploitation of voclosporin in novel formulations for treatment of autoimmune related disorders.
We have strategically developed a plan to expand our voclosporin renal franchise to include FSGS. Additionally, we are also furthering development of VOS for the treatment of DES. The advancement of these new indications, in addition to LN, represents an expansion of our pipeline and commercial opportunities.
The key elements of our corporate strategy include:
| |
• | advancing voclosporin through the AURORA Phase 3 clinical trial with anticipated completion of this trial in the fourth quarter of 2019; |
| |
• | conducting a Phase 2 proof of concept study for the additional renal indication of FSGS; and |
| |
• | conducting additional studies of VOS, while assessing mechanisms to maximize shareholder value through both clinical and business development initiatives. |
LUPUS NEPHRITIS
LN is an inflammation of the kidney caused by SLE and represents a serious manifestation of SLE. SLE is a chronic, complex and often disabling disorder. SLE is highly heterogeneous, affecting a wide range of organs and tissue systems. Unlike SLE, LN has straightforward disease measures (readily assessable and easily identified by specialty treaters) where an early response correlates with long-term outcomes, measured by proteinuria. In patients with LN, renal damage results in proteinuria and/or hematuria and a decrease in renal function as evidenced by reduced eGFR, and increased serum creatinine levels. eGFR is assessed through the Chronic Kidney Disease Epidemiology Collaboration equation. Rapid control and reduction of proteinuria in LN patients measured at six months shows a reduction in the need for dialysis at 10 years (Chen et al., Clin J. Am Soc Neph., 2008). LN can be debilitating and costly and if poorly controlled, can lead to permanent and irreversible tissue damage within the kidney. Recent literature suggests severe LN progresses to ESRD, within 15 years of diagnosis in 10%-30% of patients, thus making LN a serious and potentially life-threatening condition. SLE patients with renal damage have a 14-fold increased risk of premature death, while SLE patients with ESRD have a greater than 60-fold increased risk of premature death. Mean annual cost for patients (both direct and indirect) with SLE (with no nephritis) have been estimated to exceed US$20,000 per patient, while the mean annual cost for patients (both direct and indirect) with LN who progress to intermittent ESRD have been estimated to exceed US$60,000 per patient (Carls et al., JOEM., Volume 51, No. 1, January 2009).
DES
DES, or dry eye disease, or keratoconjunctivitis sicca, is characterized by irritation and inflammation that occurs when the eye’s tear film is compromised by reduced tear production, imbalanced tear composition, or excessive tear evaporation. The impact of DES ranges from subtle, yet constant eye irritation to significant inflammation and scarring of the eye’s surface. Discomfort and pain resulting from DES can reduce quality of life and cause difficulty reading, driving, using computers and performing daily activities. DES is a chronic disease. There are currently two FDA approved therapies for the treatment of dry eye; however, there is opportunity for potential improvement in the effectiveness by
enhancing tolerability and onset of action and alleviating the need for repetitive dosing. The disease is estimated to affect greater than 20 million people in the United States (Market Scope, 2010 Comprehensive Report on The Global Dry Eye Products Market).
FSGS
FSGS is a rare disease that attacks the kidney’s filtering units (glomeruli) causing serious scarring which leads to permanent kidney damage and even renal failure. FSGS is one of the leading causes of NS and is identified by biopsy and proteinuria. NS is a collection of signs and symptoms that indicate kidney damage, including: large amounts of protein in urine; low levels of albumin and higher than normal fat and cholesterol levels in the blood, and edema. Similar to LN, early clinical response which can be measured by reduction of proteinuria in addition to maintaining podocyte structural and functional integrity, is thought to be critical to long-term kidney health in patients with FSGS.
FSGS is likely the most common primary glomerulopathy leading to ESRD. The incidence of FSGS and ESRD due to FSGS are increasing as time goes on. Precise estimates of incidence and prevalence are difficult to determine. According to NephCure Kidney International, more than 5,400 patients are diagnosed with FSGS every year; however, this is considered an underestimate because a limited number of biopsies are performed. The number of FSGS cases are rising more than any other cause of NS and the incidence of FSGS is increasing through disease awareness and improved diagnosis. FSGS occurs more frequently in adults than in children and is most prevalent in adults 45 years or older. FSGS is most common in people of African American and Asian descent. It has been shown that the control of proteinuria is important for long-term dialysis-free survival of these patients. Currently, there are no approved therapies for FSGS in the United States or the EU.
In June 2018, we initiated a Phase 2 proof-of-concept study in FSGS, which is an open-label study of approximately 20 treatment-naive patients. The primary outcome measure for the study is the proportion of subjects achieving complete or PR at six months. Complete remission is defined as UPCR of £0.3 mg/mg, and PR is defined as 50% reduction in UPCR. This study is ongoing.
LN STANDARD OF CARE
While at Aspreva, certain members of Aurinia’s management team executed the ALMS study which established CellCept® as the current standard of care for treating LN. The ALMS study was published in 2009 in the Journal of the American Society of Nephrology and in 2011 in the New England Journal of Medicine.
The American College of Rheumatology recommends that intravenous cyclophosphamide or MMF/CellCept® be used as first-line immunosuppressive therapy for LN. Despite their use, the ALMS study showed that the vast majority of patients failed to achieve CR, and almost half failed to have a renal response at 24 weeks for both of these therapeutics. Based upon the results of the ALMS study, we believe that a better solution is needed to improve renal response rates for LN.
Despite CellCept® being the current standard of care for the treatment of LN, it remains far from adequate with fewer than 20% of patients on therapy actually achieving disease remission after six months of therapy. Data suggests that a LN patient who does not achieve rapid disease remission upon treatment is more likely to experience renal failure or require dialysis at 10 years (Chen YE, Korbet SM, Katz RS, Schwartz MM, Lewis EJ; the Collaborative Study Group. Value of a complete or partial remission in severe lupus nephritis. Clin J Am Soc Nephrol. 2008;3:46-53.). Therefore, it is critically important to achieve disease remission as quickly and as effectively as possible.
Based on available data from the AURA clinical trial, we believe that voclosporin has the potential to address critical needs for LN by controlling active disease rapidly, lowering the overall steroid burden, and doing so with a convenient oral twice-daily treatment regimen. Currently, there are no approved therapies for LN in the United States or the European Union.
MARKET POTENTIAL AND COMMERCIAL CONSIDERATIONS
We have conducted market research including claims database reviews (where available) and physician based research. Our physician research included approximately 900 rheumatologists and nephrologists across the United States, Europe and Japan to better define the potential market size, estimated pricing and treatment paradigms in those jurisdictions. Using the U.S. MarketScan® database (with approximately ~180,000,000 insured lives in the United States) there were 445,000 SLE patients in the database (between January 2006 and June 2016) based on specific SLE diagnosis codes. The National Institute of Diabetes and Digestive and Kidney Diseases estimates that up to 50% of adults with SLE are diagnosed with kidney disease at some point in their journey with lupus. Using claims database research and physician research, we believe the diagnosed range of LN patients to be approximately 125,000 to 180,000 in the United States and 150,000 to 215,000 for Europe and Japan combined. According to our research, in the United States, Europe and Japan, one in five LN patients are thought to be undiagnosed due to referring physicians being inefficient and inaccurate in diagnosing the condition.
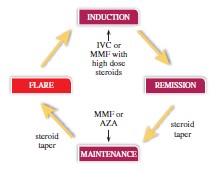
Similar to other autoimmune disorders, LN is a flaring and remitting disease. The destructive disease cycle people with LN go through is depicted below. The disease cycles from being in remission to being in flare, achieving PR and being back in remission. Treatment objectives between LN and other autoimmune diseases are remarkably similar. In other autoimmune conditions such as Multiple Sclerosis, Crohn’s, Rheumatoid Arthritis and SLE, physicians’ goals are to induce/maintain a remission of disease, decrease frequency of hospital or ambulatory care visits and limit long term disability. In LN specifically, physicians are trying to avoid further kidney damage, dialysis, renal transplantation, and death. According to a physician survey, the frequency of LN flares amongst treated patients was approximately every 14 months across the United States and Europe. The ability to get patients into remission quickly correlates with better long-term kidney outcomes as noted above.
The population of people with LN will be in different cycles of their disease at any one time. Physicians currently use existing LN standard of care including immunosuppressants and high dose steroids to treat people with LN throughout the disease cycles including induction and maintenance phases. By studying voclosporin on top of an existing standard of care we are not seeking to displace current accepted treatment patterns. We feel that being additive to an existing standard of care in addition to the product being administered orally versus via infusion or injection can support a more rapid market adoption if approved.
Current annualized pricing (based on wholesale acquisition costs published by AnalySource® Reprinted with permission by First Databank, Inc. (All rights reserved. © 2018) for the treatments of other more prevalent autoimmune conditions such as Multiple Sclerosis, Crohn’s, Rheumatoid Arthritis and SLE ranges from US$45,000 to US$100,000 in the United States. Wholesale acquisition cost is the manufacturer’s published catalog or list price for a drug product to wholesalers and may not reflect actual prices paid after any rebates/ discounts. We have conducted pricing research that studied a similar pricing range with payers and physicians and believe that pricing in this range may be achievable for voclosporin in the United States. Pricing for other autoimmune conditions are lower in Europe and Japan than they are in the United States driven by the specific country’s pricing and reimbursement processes. We expect that will be the case for voclosporin.
VOCLOSPORIN BACKGROUND
Voclosporin mechanism of action
Voclosporin reversibly inhibits immunocompetent lymphocytes, particularly T-Lymphocytes in the G0 and G1 phase of the cell-cycle, and also reversibly inhibits the production and release of lymphokines. Through a number of processes voclosporin inhibits and prevents the activation of various transcription factors necessary for the induction of cytokine genes during T-cell activation. It is believed that the inhibition of activation of T-cells will have a positive modulatory effect in the treatment of LN. In addition to these immunologic impacts recent data suggests that CNIs have another subtle but important impact on the structural integrity of the podocytes (Faul C, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of CsA. Nat Med. 2008 Sep;14(9):931-8. doi: 10.1038/nm.1857). This data suggests that inhibition of calcineurin in patients with autoimmune kidney diseases helps stabilize the cellular actin-cytoskeleton of the podocytes thus having a structural impact on the podocyte and the subsequent leakage of protein into the urine, which is a key marker of patients suffering from LN.
Potential voclosporin clinical benefits
We believe that voclosporin has shown a number of key potential clinical benefits over the existing commercially available CNIs (tacrolimus & CsA). Firstly, CNI assay results have indicated that voclosporin is approximately four times more potent than its parent molecule CsA, which would indicate an ability to give less drug and produce fewer potentially harmful metabolites. Secondly, CsA inhibits the enterohepatic recirculation of MPA, the active metabolite of MMF. The net effect of co-administration of CsA with MMF is reduced MPA systemic exposure by as much as 50% (D. Cattaneo et al. American Journal of Transplantation, 2005:12(5);2937-2944.). This drug interaction has not been observed with voclosporin and it is not expected that MPA blood exposure levels will be reduced with voclosporin co-administration. This is an important fact to consider as most patients being treated with voclosporin for LN will already be taking MMF. Furthermore, PK-PD analysis indicate lower PK-PD variability for voclosporin versus tacrolimus or CsA, to the extent that we believe flat-dosing can be achieved for voclosporin. The currently available CNIs require extensive therapeutic drug monitoring which can often be costly, confusing and time consuming for treating physicians.
In a head-to-head study comparing voclosporin against CsA in the treatment of psoriasis, CsA was shown to cause significant increases in lipid levels as compared to voclosporin. The difference was statistically significant. This is important considering most lupus patients die of cardiovascular disease. In another study comparing voclosporin against tacrolimus in patients undergoing renal transplantation, the voclosporin group experienced a statistically significantly lower incidence of glucose intolerance and diabetes than tacrolimus treated patients. Additionally, in the Japanese tacrolimus study that led to the approval of this drug in Japan, almost 15% of tacrolimus patients experienced glucose intolerance (Miyasaka N, Kawai S, Hashimoto H. Efficacy and safety of tacrolimus for lupus nephritis: a placebo-controlled double-blind multicenter study. Mod Rheumatol. 2009;19(6):606-15. Epub 2009 Aug 18). This is a major limitation for physicians wanting to use this agent in lupus and is a well described side effect of tacrolimus.
We believe that voclosporin can be differentiated from the older CNIs and thus possess a unique position in the market as it relates to inducing remission in patients suffering from LN.
Scientific Rationale for Treatment of LN with voclosporin
While SLE is a highly heterogeneous autoimmune disease (often with multiple organ and immune system involvement), LN has straightforward disease outcomes. T-cell mediated immune response is an important feature of the pathogenesis of LN while the podocyte injury that occurs in conjunction with the ongoing immune insult in the kidney is an important factor in the clinical presentation of the disease. An early response in LN correlates with long-term outcomes and is clearly measured by proteinuria.
The use of voclosporin in combination with the current standard of care for the treatment of LN provides a novel approach to treating this disease (similar to the standard approach in preventing kidney transplant rejection). Voclosporin has shown to have potent effects on T-cell activation leading to its immunomodulatory effects. Additionally, recent evidence suggests that inhibition of calcineurin has direct physical impacts on the podocytes within the kidney. Inhibition of calcineurin within the podocytes can prevent the dephosphorylation of synaptopodin which in turn inhibits the degradation of the actin cytoskeleton within the podocyte. This process is expected to have a direct impact on the levels of protein in the urine which is a key marker of LN disease activity.
Voclosporin Development History
More than 2,400 patients have been dosed with voclosporin in clinical trials including studies where voclosporin was compared to placebo or active control. The safety and tolerability profile of the drug therefore is well characterized. Phase 2 or later clinical studies that have been completed include studies in the following indications:
Psoriasis: Two Phase 3 clinical studies in patients with moderate to severe psoriasis have been completed. The primary efficacy endpoint in both studies was a reduction in Psoriasis Area and Severity Index, which is a common measure of psoriasis disease severity. The first study treatment with voclosporin resulted in statistically significantly greater success rates than treatment with placebo by the twelfth week. In a second study comparing voclosporin against cyclosporine, the drug was not shown to be statistically non-inferior to cyclosporine in terms of efficacy; however, voclosporin proved superior in terms of limiting elevations in hyperlipidemia. Due to the evolving psoriasis market dynamics and the changing standard of care for the treatment of this disease, we have decided not to pursue further Phase 3 development.
Renal Transplantation: A Phase 2b clinical trial in de novo renal transplant recipients was completed. Study ISA05-01, the PROMISE Study (Busque S, Cantarovich M, Mulgaonkar S, Gaston R, Gaber AO, Mayo PR, et al; PROMISE Investigators. The PROMISE study: a phase 2b multicenter study of voclosporin (ISA247) versus tacrolimus in de novo kidney transplantation. Am J Transplant. 2011 Dec;11(12):2675-84) was a six-month study with a six-month extension comparing voclosporin directly against tacrolimus on a background of MMF and corticosteroids. Voclosporin was shown to be equivalent in efficacy, but superior to tacrolimus with respect to the incidence of new onset diabetes after transplantation. In 2010, tacrolimus lost its exclusivity in most world markets and as a result, the competitive pricing environment for voclosporin for this indication has come into question. Additionally, the more expensive development timelines for this indication has made it a less attractive business proposition as compared to the LN indication, even when considering the fact that a special protocol assessment has been agreed to by the FDA for this indication.
Uveitis: Multiple studies in various forms of non-infectious uveitis were completed by Lux, one of our former licensees, indicating mixed efficacy. In all but one of the studies, completed by the licensee, an impact on disease activity was shown in the voclosporin group. However, achievement of the primary end-points in multiple studies could not be shown. Uveitis is a notoriously difficult disease to study due to the heterogeneity of the patient population and the lack of validated clinical end-points. However, in all of the uveitis studies completed, the safety results were consistent, and the drug was well tolerated. We successfully terminated our licensing agreement with Lux on February 27, 2014. In conjunction with this termination we have retained a portfolio of additional patents that Lux had been prosecuting that are focused on delivering effective concentrations of voclosporin to various ocular tissues.
RECENT DEVELOPMENTS
PATENT NOTICE OF ALLOWANCE
On February 25, 2019, we announced that we had received a Notice of Allowance from the USPTO for claims directed at our novel voclosporin dosing protocol for LN (U.S. patent application 15/835,219, entitled "PROTOCOL FOR TREATMENT OF LUPUS NEPHRITIS”).
The allowed claims broadly cover the novel voclosporin individualized flat-dosed pharmacodynamic treatment protocol adhered to and required in both the previously reported Phase 2 AURA-LV trial and our ongoing Phase 3 confirmatory AURORA trial. Notably, the allowed claims cover a method of modifying the dose of voclosporin in patients with LN based on patient specific pharmacodynamic parameters.
This Notice of Allowance concludes a substantive examination of the patent application at the USPTO, and after administrative processes are completed and fees are paid, is expected to result in the issuance of a U.S. patent with a term extending to December 2037. If the FDA approves the use of voclosporin for LN and the label for such use follows the dosing protocol under the Notice of Allowance, the issuance of this patent will expand the scope of intellectual property protection for voclosporin, which already includes robust manufacturing, formulation, synthesis and composition of matter patents.
We have also filed for protection of this subject matter under the PCT and have the option of applying for similar protection in the member countries thereof. This may lead to the granting of corresponding claims in the treaty countries which include all the major global pharmaceutical markets.
DES
On January 22, 2019 we released results for our exploratory Phase 2a head-to-head study evaluating the efficacy, safety and tolerability of VOS versus Restasis® (cyclosporine ophthalmic emulsion) 0.05% for the treatment of DES. The study was initiated in July of 2018 and full enrollment was achieved in the fourth quarter of 2018. We believe CNIs are a mainstay of treatment for DES. The goal of this program is to develop a best-in class treatment option.
In this exploratory Phase 2a study:
| |
• | VOS showed statistical superiority to Restasis® on FDA-accepted objective signs of DES |
| |
▪ | 42.9% of VOS subjects vs 18.4% of Restasis® subjects (p=.0055) demonstrated ≥ 10mm improvement in STT at Week 4 |
| |
▪ | Primary endpoint of drop discomfort at 1-minute on Day 1 showed no statistical difference between VOS and Restasis®, as both exhibited low drop discomfort scores, and both drugs were well-tolerated. |
On the key pre-specified secondary endpoints of Schirmer Tear Test/STT (an objective measure of tear production), and Fluorescein Corneal Staining/FCS (an objective measure of structural damage to the cornea), which are FDA-accepted efficacy endpoints, VOS showed rapid and statistically significant improvements over Restasis® at Week 4 (STT: p=.0051; FCS: p=.0003).
This 100-patient, double-masked, head-to-head study was designed to evaluate the efficacy, safety and tolerability of VOS versus Restasis® in subjects with DES. Both arms of the study received either VOS or Restasis® (1:1) administered twice daily, in both eyes, for 28 days. Key pre-specified secondary endpoints, which are FDA-accepted endpoints, include STT, FCS, and assessments of dry eye symptoms. Improvements in STT and FCS are considered by regulators to be two of the most clinically meaningful measures of efficacy in this disease.
|
| | | |
4-Week Pre-Specified Efficacy Endpoints (Signs)* | VOS | Restasis® | p-value vs. Restasis® |
Schirmer Tear Test (STT) (mm LS mean increase from baseline) | 8.6 | 3.3 | .0051 |
% of subjects showing ≥ 10mm improvement in STT (basis of FDA approval for other CNIs and an improvement is considered to be clinically significant) | 42.9% | 18.4% | .0055 |
Fluorescein Corneal Staining (FCS) (reduction in staining is clinically significant) | -2.2 | -0.2 | .0003 |
* worst eye
Both treatment arms also demonstrated substantial and statistically significant improvements on the Symptom Assessment in Dry Eye (SANDE) score from baseline to Week 4.
No serious adverse events (SAE) were reported in the study, and there were no unexpected safety signals. All adverse events (AEs) were mild to moderate and the majority of patients had no AEs. There were five more patients with mild to moderate AEs in the VOS vs Restasis arm which were typical of complaints from DES patients.
Based on this data, we plan to aggressively advance VOS for the treatment of DES. Our pursuit of further development of VOS provides the Company with an enhanced pipeline that further capitalizes on the differentiating features of voclosporin and positions us for substantial growth and measured diversification.
VOS, had previously shown evidence of efficacy in our partnered canine studies and in a small human Phase 1 study (n=35), supporting its development for the treatment of DES. Completed preclinical and human Phase 1b studies using our nanomicellar VOS formulation have shown encouraging results in terms of delivery of active drug to the target tissues of the eye. The nanomicellar formulation enables high concentrations of voclosporin to be incorporated into a preservative-free solution for local delivery to the ocular surface. This has been shown to potentially improve efficacy, dosing frequency and tolerability versus the current treatments for DES. We therefore believe VOS has a differentiated product profile with long patent life that has the potential to compete in the multi-billion-dollar human prescription dry eye market.
Animal safety toxicology studies were previously completed in rabbit and dog models, and additional animal safety toxicology studies are either being currently conducted or in the planning stage for 2019.
NOVEMBER 2018 ATM
On November 30, 2018 we entered into an open market sale agreement with Jefferies LLC pursuant to which Aurinia would be able to, from time to time, sell, through at-the-market (“ATM”) offerings, Common Shares that would have an aggregate offering price of up to US$30 million. Aurinia filed a prospectus supplement with securities regulatory authorities in Canada in the provinces of British Columbia, Alberta and Ontario, and with the United States Securities and Exchange Commission, which supplemented Aurinia’s short form base shelf prospectus dated March 26, 2018, and Aurinia’s shelf registration statement on Form F-10 dated March 26, 2018, declared effective on March 29, 2018.
Subsequent to year-end, we sold 4.61 million Common Shares and received gross proceeds of US$30 million at a weighted average price of US$6.55 pursuant this agreement. We incurred share issue costs of US$1.17 million including a 3% commission of US$900,000 and professional and filing fees of US$270,000 directly related to the ATM offering. Sales in the ATM offering were only conducted in the United States through NASDAQ at market prices.
THREE YEAR HISTORY
CLINICAL AND CORPORATE DEVELOPMENTS IN 2018
AURORA clinical trial
We achieved a significant milestone on September 25, 2018 with the completion of enrollment for our AURORA Phase 3 clinical trial. The target enrollment of 324 patients was surpassed due to high patient demand with 358 LN patients randomized in sites across 27 countries. AURORA is a 56-week trial (52-week primary endpoint and a four-week follow-up period). We expect to have top-line data for this trial in late 2019.
We believe the totality of data from both the AURORA and AURA clinical trials can potentially serve as the basis for a NDA submission with the FDA following a successful completion of the AURORA clinical trial. Under voclosporin’s fast-track designation we intend to utilize a rolling NDA process which will allow us to begin the submission process following a positive pre-NDA meeting with the FDA, which we anticipate will occur in the first quarter of 2020. To that end we are actively preparing the non-clinical and Chemistry, Manufacturing and Controls modules required for the NDA submission. Our current plan is to complete the NDA submission, including the clinical module, in the second quarter of 2020 and therefore we do not expect any delay in our originally planned regulatory timelines.
The AURORA clinical trial is a global double-blind, placebo-controlled study, (designed with target enrollment of 324 patients) to evaluate whether voclosporin added to background therapy of CellCept®/MMF can increase overall renal response rates in the presence of low dose steroids.
Patients were randomized 1:1 to either of: (i) 23.7 mg voclosporin BID and MMF, or (ii) MMF and placebo, with both arms receiving a rapid oral corticosteroid taper. As in the AURA clinical trial, the study population in AURORA is comprised of patients with biopsy proven active LN who will be evaluated on the primary efficacy endpoint of CR, or renal response, at 52 weeks, a composite which includes:
| |
• | normal, stable renal function (≥60 mL/min/1.73m2 or no confirmed decrease from baseline in eGFR of >20%); |
| |
• | presence of sustained, low dose steroids (≤10mg prednisone from week 44-52) and; |
| |
• | no administration of rescue medications. |
Patients completing the AURORA trial have the option to roll over into a 104-week blinded extension trial (the "AURORA 2 extension trial"). During the second quarter ended June 30, 2018, the first patients commenced rolling over into the AURORA 2 extension trial. Enrollment in this study continues to increase as additional patients complete AURORA. The data from the AURORA 2 extension trial will allow us to assess the long-term benefit/risk of voclosporin in LN patients, however, this study is not a requirement for potential regulatory approval for voclosporin. Data from the AURORA 2 extension trial assessing long-term outcomes in LN patients should be valuable in a post-marketing setting and for future interactions with various regulatory authorities.
In order to enhance and complete the clinical dossier, we commenced a confirmatory drug-drug interaction study between voclosporin and MMF in the second half of 2018. Legacy CNIs, CsA, impact MMF concentrations, and our goal with this short study is to confirm the insignificant impact of voclosporin upon MMF concentrations that were previously seen in a renal transplant study. We are conducting the drug-drug interaction study with SLE patients and are currently in the process of enrolling patients with the study expected to be completed in 2019. In this study, patients will be monitored for a period of two weeks. We believe the results of this study will add to our knowledge of voclosporin in a MMT approach and should have no impact on our submission time-line or the potential approval of voclosporin.
We also plan to evaluate voclosporin in pediatric patients after a potential FDA approval of an indication for adults with LN.
New Voclosporin Indication - FSGS
Similar to LN, integrity of the podocyte is a key feature of disease progression in FSGS. The disease has straightforward disease outcomes where an early clinical response correlates with long-term outcomes, measured by proteinuria. Based on our clinical data in LN which demonstrated that voclosporin decreased proteinuria, we believe voclosporin has the potential to benefit patients with FSGS. Our clinical data in LN demonstrated that voclosporin decreased proteinuria. Furthermore, voclosporin appears to demonstrate a more predictable pharmacology and an improved lipid and metabolic profile over legacy calcineurin inhibitors, which have shown efficacy in treating autoimmune disorders similar to those we are targeting.
We submitted our IND to the FDA in the first quarter of 2018. We received agreement from the FDA with regards to the guidance we provided on this study and the IND is now active. Our Phase 2 proof-of-concept study in FSGS which is an open-label study of approximately 20 treatment-naive patients was initiated in June 2018. As we are essentially enrolling newly diagnosed patients and this is a rare disease, enrollment is slower than originally expected. We believe enrollment could take up to an additional twelve months from the current date, however, we plan to have interim data readouts throughout the course of the study, once sufficient patients are enrolled. As we have been focused on LN, expanding our scope to include other proteinuric renal diseases is synergistic with our current strategy and long-term vision.
Corporate Developments
On February 21, 2018 we appointed Michael Hayden, CM, OBC, MB, ChB, PhD, FRCP (C), FRSC to our Board. Dr. Hayden was most recently the President of Global R&D and CSO at Teva Pharmaceutical Industries Ltd. Dr. Hayden is the co-founder of three biotechnology companies,
including Aspreva, and currently sits on several boards. Dr. Hayden is a celebrated researcher, having focused his research primarily on genetic diseases.
On February 7, 2018 we appointed Joseph P. "Jay" Hagan to our Board. Mr. Hagan is currently the President and CEO of Regulus Therapeutics, having previously held the positions of COO, Principal Financial Officer and Principal Accounting Officer.
We announced on November 8, 2018 that Richard M. Glickman, Aurinia's Chairman and CEO, intends to retire from his position once a suitable replacement is identified and appointed. The Board has retained an executive search firm and initiated a search for his successor. Under his direction, the Company has delivered on its key milestones and evolved into a patient-centric, late-stage clinical company with investigational drugs addressing multiple indications across the global immunology market.
CLINICAL AND CORPORATE DEVELOPMENTS IN 2017
Initiation of AURORA clinical trial
We achieved a significant milestone in the second quarter of 2017 with the initiation of our single, AURORA clinical trial with patients randomized on active treatment.
We believe the totality of data from both the AURORA and AURA clinical trials, if the AURORA results confirm the AURA data, can potentially serve as the basis for a NDA submission following a successful completion of the AURORA clinical trial.
AURA-LV 48-Week Results
On April 20, 2017, we presented in-depth 48-week results from our global AURA clinical trial in LN during the late-breaking session at National Kidney Foundation 2017 Spring Clinical Meetings in Orlando, Florida. These were updated results from the top-line remission rate results announced on March 1, 2017 and are summarized in the table below. In addition to the trial meeting its CR and PR endpoints at 48 weeks, all pre-specified secondary endpoints that had been analyzed to April 20, 2017 were also met at 48 weeks. These pre-specified endpoints included: time to CR and PR (speed of remission); reduction in SLEDAI score; and reduction in UPCR over the 48-week treatment period.
Each arm of the trial included the current standard of care of MMF as background therapy and a rapid steroid taper to 5mg/day by week 8 and 2.5mg/day by week 16. Both doses of voclosporin at 48 weeks demonstrated continued improvement over the control group across multiple dimensions. Notably, the voclosporin groups demonstrated statistically significantly improved speed and rates of CR and PR. Of the patients that achieved CR at 24 weeks, in the low-dose voclosporin group, 100% remained in CR at 48 weeks, which demonstrates durability of clinical response. Proteinuria levels and reduction in SLEDAI scores, which include non-renal measures of lupus activity, also continued to significantly separate over time versus the control group.
The 24 and 48-week efficacy results are summarized below:
|
| | | | | |
Endpoint | Treatment | 24 weeks | P-value* | 48 weeks | P-value* |
Complete Remission (CR) | 23.7mg VCS BID | 33% | p=.045 | 49% | p<.001 |
39.5mg VCS BID | 27% | p=.204 | 40% | p=.026 |
Control Arm | 19% | NA | 24% | NA |
Partial Remission (PR) | 23.7mg VCS BID | 70% | p=.007 | 68% | p=.007 |
39.5mg VCS BID | 66% | p=.024 | 72% | p=.002 |
Control Arm | 49% | NA | 48% | NA |
Time to CR (TTCR) [median] | 23.7mg VCS BID | 19.7 weeks | p<.001 | 19.7 weeks | p<.001 |
39.5mg VCS BID | 23.4 weeks | p=.001 | 23.4 weeks | p<.001 |
Control Arm | NA | NA | NA | NA |
Time to PR (TTPR) [median] | 23.7mg VCS BID | 4.1 weeks | p=.002 | 4.3 weeks | p=.005 |
39.5mg VCS BID | 4.4 weeks | P=.003 | 4.4 weeks | p=.002 |
Control Arm | 6.6 weeks | NA | 6.6 weeks | NA |
SLEDAI Reduction (non-renal lupus) | 23.7mg VCS BID | -6.3 | p=.003 | -7.9 | p<.001 |
39.5mg VCS BID | -7.1 | p=.003 | -8.3 | p<.001 |
Control Arm | -4.5 | NA | -5.3 | NA |
Reduction in UPCR | 23.7mg VCS BID | -3.769 mg/mg | p<.001 | -3.998 mg/mg | p<.001 |
39.5mg VCS BID | -2.792 mg/mg | p=.006 | -2.993 mg/mg | p=.008 |
Control Arm | -2.216 mg/mg | NA | -2.384 mg/mg | NA |
The results of the AURA clinical trial at 48 weeks demonstrate the highest CR rate of any global LN study of which we are aware, although we note that the criteria to measure remission differs among various studies. The below chart compares the results of the AURA clinical trial vs. the other global LN studies of which we are aware.
|
| | | | |
Name of Global Study | Number of weeks | Criteria to Measure Remission and Response Rate | Results |
Efficacy and Safety of Ocrelizumab in Active Proliferative LN | 48 weeks | UP:CR(gm/gm) < .5 SCr ≤ 25% increase from baseline Steroid taper (not enforced) | Control = 34.7% LD OCR = 42.7% (NS) HD OCR = 32.5% (NS) |
Mycophenolate Mofetil versus Cyclophosphamide for Induction Treatment of LN | 24 weeks | UP:CR(gm/gm) ≤ .5 Normal eGFR Normal Urinalysis Steroid taper (not enforced) | MMF = 8.6% (NS) IVC = 8.1% (NS) |
Efficacy and Safety of Abatacept in LN | 52 weeks | UP:CR(gm/gm) ≤ .26 eGFR within 10% of screening/baseline Normal Urinalysis Criteria to be met on 2 successive visits No mandated steroid taper | Control = 8.0% LD ABT = 11.1% (NS) HD ABT = 9.1% (NS) |
AURA-LV: Aurinia Urine Protein Reduction in Active LN Study | 24 and 48 weeks | UP:CR(gm/gm) ≤ .5 No decrease in eGFR ≥ 20% No use of rescue medications Forced steroid taper | 24 weeks Control = 19.3% LD Voc=32.6% (p=.045) HD Voc = 27.3% (NS) | 48 weeks Control = 23.9% LD Voc = 49.4% (p<.001) HD Voc = 39.8% (p=.026) |
No new safety signals were observed with the use of voclosporin in LN patients, and voclosporin was well-tolerated over a 48-week period. The overall safety profile is consistent with the expectations for the class of drug, the patient population and concomitant therapies. Thirteen (13) deaths were reported during the AURA clinical trial, a pattern which is consistent with other global active LN studies. Eleven (11) of the thirteen (13) deaths occurred at sites with compromised access to standard of care, and patients who died had a statistically different clinical baseline picture, demonstrating a more severe form of LN, potential comorbid conditions, and poor nutrition. Furthermore, in the voclosporin arms, the renal function as measured by corrected eGFR was stable and not significantly different from the control arm after 48 weeks of treatment. Mean blood pressure was also similar between all treatment groups.
A summary of TEAEs, study withdrawals and drug discontinuations are below, which are consistent with other clinical trials evaluating immunosuppressive therapies.
|
| | | |
TEAEs, Drug Discontinuation & Study Withdrawals | Control N=88 n (%) | VCS 23.7 mg BID N=89 n (%) | VCS 39.5mg BID N=88 n (%) |
Any TEAE | 78 (88.6) | 82 (92.1) | 85 (96.6) |
Any Serious TEAE | 17 (19.3) | 25 (28.1) | 22 (25.0) |
Any TEAE with Outcome of Death1 | 4 (4.5) | 10 (11.2) | 2 (2.3) |
Any Treatment-Related TEAE | 15 (17.0) | 45 (50.6) | 55 (62.5) |
Any Serious Treatment-Related TEAE | 1 (1.1) | 4 (4.5) | 7 (8.0) |
Any AE leading to study drug discontinuation | 9 (10.2) | 16 (18.0) | 14 (15.9) |
Any AE leading to study drug discontinuation (excluding deaths) | 8 (9.1) | 11 (12.4) | 13 (14.8) |
Study Withdrawals | 18 (20) | 16 (18.0) | 8 (9.1) |
1. Data includes three placebo-randomized subjects that died post-study completion.
On June 4, 2017 and June 14, 2017, we presented additional data from the AURA trial in LN during ERA-EDTA 2017 and EULAR 2017.
As previously reported, treatment with low dose voclosporin showed statistically improved efficacy over the control arm at 24 and 48 weeks. The data presented at ERA-EDTA demonstrated this improved efficacy was attained while maintaining stable serum magnesium, potassium and blood pressure levels. Well-known side effects with other calcineurin inhibitors at their effective dose include hypomagnesemia and hyperkalemia, which are associated with renal impairment and require monitoring or intervention.
The data presented at EULAR 2017 demonstrated that over the course of the 48-week trial, patients on voclosporin stayed in remission approximately twice the amount of time as those in the control group.
The analysis of additional data after April 20, 2017 identified that two non-key secondary endpoints: urine sediment, which describes analysis of active urinary sediment at each visit; and comparison of C3 and C4 levels between study arms, did not demonstrate statistical significance between arms. The urine sediment endpoint was not statistically different as there was too few data to demonstrate a difference. C3 and C4 levels are non-specific markers of general lupus disease activity. Rises in C3 and C4 were seen in all arms indicating disease improvement though no significant difference was observed between treatment arms.
To summarize, in addition to the trial meeting its CR and PR endpoints at 48 weeks, all key pre-specified secondary endpoints were also met at 48 weeks.
AURORA to serve as basis for regulatory submissions in major markets-US, Europe, and Japan
On April 6, 2017, we announced the outcome of discussions with both the EMA and the PMDA in Japan regarding the development of voclosporin for the treatment of active LN. Pursuant to these discussions, we believe that the confirmatory data that can be generated from the AURORA clinical trial and the recently completed AURA clinical trial should support regulatory submissions in the US, Europe and Japan.
48-week data from open-label AURION clinical trial
On March 27, 2017, we presented the 48-week results from the open-label AURION clinical trial at the 12th International Congress on Systemic Lupus Erythematosus and the 7th Asian Congress on Autoimmunity jointly in Melbourne, Australia.
The trial successfully achieved its primary objective by demonstrating that early biomarker response in active LN patients can be a significant predictor of renal response at 24 and 48 weeks. In the per protocol analysis at 48 weeks, 71% of subjects (n=5/7) on treatment remain in CR as measured by a UPCR of ≤ 0.5mg/mg, eGFR within 20% of baseline and concomitant steroid dose of <5mg/day. A 25% reduction in UPCR at week eight was found to be highly predictive of achieving renal response at 24 and 48 weeks. Conversely, if C3 and C4 do not normalize by week 8, then a renal response at week 24 and 48 is highly unlikely. Anti-dsDNA was not found to be a useful biomarker in predicting long-term response in LN patients.
No new safety signals were observed with the use of voclosporin in LN patients; voclosporin was well-tolerated, and the safety profile was consistent with other immunomodulators. A total of three subjects were discontinued prior to 48 weeks due to lupus related complications or investigator discretion.
Results from AURION demonstrated that an early UPCR reduction of 25% is the best predictor of renal response at 24 and 48 weeks. In addition, the use of C3 or C4 improves the precision of predicting if a patient will achieve a clinical response. This exploratory study is supportive of the successful AURA clinical trial.
Each arm of the trial included the current standard of care of MMF as background therapy and a forced steroid taper to 5mg/day by week 8 and 2.5mg/day by week 16.
Results from Japanese Phase 1 Ethno-bridging Study for Voclosporin
On February 14, 2017, we announced the results of a supportive Phase 1 safety PK-PD study in healthy Japanese patients which supports further development of voclosporin in this patient population. Based on evaluations comparing the Japanese ethno-bridging data vs. previous PK and PD studies in non-Japanese patients, voclosporin demonstrated no statistically significant differences in exposure with respect to Area Under the Curve measurements. Furthermore, the PK parameters in Japanese patients were generally consistent with previously evaluated PK parameters in non-Japanese volunteers. There were no unusual or unexpected safety signals in the study.
March 20, 2017 Offering
On March 20, 2017, we completed an underwritten public offering of 25.64 million Common Shares, which included 3.35 million Common Shares issued pursuant to the full exercise of the underwriters’ overallotment option to purchase additional Common Shares (the "March Offering"). The Common Shares were sold at a public offering price of US$6.75 per share. The gross proceeds from the March Offering were US$173.10 million before deducting the 6% underwriting commission and other offering expenses which totaled US$10.78 million. Leerink Partners LLC and Cantor Fitzgerald & Co. acted as joint book-running managers for the March Offering. The March Offering was made pursuant to a U.S. registration statement on Form F-10, declared effective by the SEC on November 5, 2015 (the "Registration Statement"), and the Company’s existing Canadian short form base shelf prospectus (the "2015 Base Shelf Prospectus") dated October 16, 2015. The prospectus supplements relating to the Offering (together with the 2015 Base Shelf Prospectus and the Registration Statement) were filed with the securities commissions in the provinces of British Columbia, Alberta and Ontario in Canada, and with the SEC in the United States.
Changes to Board and Management
On February 6, 2017, we appointed Dr. Richard M. Glickman LLD (Hon), our founder and Chairman of the Board, as our Chairman and CEO. The Board accepted the resignation of Charles Rowland as CEO and an executive member of the Board.
On May 9, 2017, we appointed George M. Milne Jr., PhD to the Board. Prior to his retirement, Dr. Milne served as Executive Vice President of Global Research and Development and President of Worldwide Strategic and Operations Management at Pfizer. Dr. Milne serves on multiple corporate boards including Charles River Laboratories where he is the lead director and Amylyx Pharmaceuticals and is a Venture Partner at Radius Ventures. On May 8, 2017, Dr. Gregory Ayers resigned from the Board.
On April 17, 2017, we hired Simrat Randhawa MD, MBA, as Head of Medical Affairs. Simrat brings over 20 years of experience to Aurinia across clinical practice, medical affairs and business development. For the past 10 years, he has held a number of senior leadership roles in commercial and medical affairs within large and small pharmaceutical companies. During this time, Simrat served as the medical lead for Novartis' Multiple Sclerosis (MS) franchise, where he played an integral role in establishing Gilenya® as the first oral therapy for the treatment of Relapsing MS. Most recently he was the global medical affairs lead at BioMarin Pharmaceuticals for MPS, Duchenne Muscular Dystrophy and Hemophilia
On July 3, 2017, we hired Erik Eglite, DPM, JD, MBA as Senior Vice President, General Counsel & Chief Corporate Compliance Officer. Prior to joining Aurinia, Erik was Vice President, Chief Compliance Officer and Corporate Counsel for Marathon Pharmaceuticals and Vice President, Chief Compliance Officer and Corporate Counsel for Lundbeck Pharmaceuticals. Prior to that, he was Vice President, Chief Compliance Officer and Corporate Counsel for Ovation Pharmaceuticals and Global Chief Compliance Officer, Corporate Counsel for Aspreva Pharmaceuticals. Erik has been involved with the clinical development, launch and commercialization of 12 drugs and drug programs. He is also a licensed podiatric physician and surgeon.
Termination of Paladin Agreements
Effective December 28, 2017, we terminated the License Agreement dated June 18, 2009 between Paladin and the Company (as amended). Concurrent with the termination of the License Agreement, under the terms of the R&D Agreement dated June 18, 2009, between Paladin and the Company (as amended), the R&D Agreement also terminated effective December 28, 2017.
CLINICAL AND CORPORATE DEVELOPMENTS IN 2016
FDA End of Phase 2 Meeting and Plans for Single LN Phase 3 Clinical Trial
On November 2, 2016, we announced the FDA’s preference for a single LN Phase 3 clinical trial for voclosporin in the treatment of LN and our plans and expectations for the AURORA clinical trial. A further description of the AURORA clinical trial is set out under the headings "Three Year History - Clinical and Corporate Developments in 2018 - AURORA clinical trial" and "Three Year History - Clinical and Corporate Developments in 2017 - Initiation of AURORA clinical trial".
AURION Clinical Trial Update
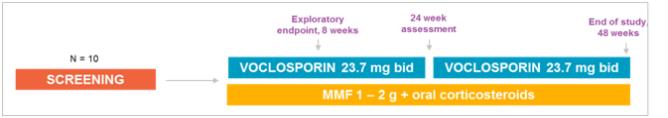
The AURION trial was a single-arm, twin center, exploratory study assessing the predictive value of an early reduction in proteinuria in subjects receiving 23.7 mg of voclosporin BID with the current standard of care in patients with active LN. The primary objective of the AURION clinical trial was to examine biomarkers of disease activity at eight weeks and their ability to predict response at 24 and 48 weeks.
Study Design:
The primary analysis is the number of patients achieving each of the following biomarkers and the number of these patients who go on to achieve week 24 or week 48 remission.
Biomarkers:
| |
• | 25% reduction in UPCR at eight weeks; |
| |
• | C3 complement normalization at 8 weeks; |
| |
• | C4 complement normalization at 8 weeks; and |
| |
• | Anti-dsDNA normalization at eight weeks. |
The secondary analysis includes the 24 and 48-week outcomes, markers of SLE and PK-PD of voclosporin.
On October 6, 2016, we announced 24-week data in all 10 patients from the AURION clinical trial, an open-label exploratory study to assess the short-term predictors of response using voclosporin (23.7 mg BID) in combination with MMF and oral corticosteroids in patients with active LN. The data was presented by Robert Huizinga, Vice President of Clinical Affairs at Aurinia Pharmaceuticals at the 10th Annual European Lupus Meeting in Venice, Italy.
The primary objective of the trial is to examine biomarkers of disease activity at eight weeks and their ability to predict response at 24 and 48 weeks.
In this trial, 70% (7/10) of patients achieved CR at 24 weeks as measured by a UPCR of 0.5mg/mg, eGFR within 20% of baseline and concomitant steroid dose of <5 mg/day. Of the 10 patients that achieved a reduction of UPCR of 25% at 8 weeks, 80% were responders (50% reduction in UPCR over baseline) at 24 weeks and 70% were in CR at 24 weeks, proteinuria levels decreased by a mean of 61% from baseline through the
first 24 weeks of the study. In addition, inflammatory markers such as C3, C4 and Anti-dsDNA all continued to normalize to 24 weeks. Voclosporin was well-tolerated with no unexpected safety signals observed. Renal function, as measured by eGFR, also remained stable over the 24 weeks. We believe that the results of the AURION clinical trial supports the use of the 23.7 mg twice daily dose in further studies.
Details of the results are below:
|
| | | | | | | | | |
Patient# | Attained ≥25% reduction in UPCR at 8 weeks | | Attained PR* at 8 weeks | | Attained PR* at 24 weeks | | Attained CR at 8 weeks | | Attained CR at 24 weeks |
1 | Y | | Y | | Y | | Y | | Y |
2 | Y | | Y | | Y | | Y | | Y |
3 | Y | | Y | | Y | | N | | N |
4 | Y | | N | | N | | N | | N |
5 | Y | | Y | | Y | | Y | | Y |
6 | Y | | Y | | Y | | Y | | Y |
7 | Y | | N | | N | | N | | N |
8 | Y | | Y | | Y | | Y | | Y |
9 | Y | | N | | Y | | N | | Y |
10 | Y | | Y | | Y | | N | | Y |
TOTALS: | 100% (10/10) | | 70% (7/10) | | 80% (8/10) | | 50% (5/10) | | 70% (7/10) |
| |
* | Retrospectively defined by ≥50% reduction in UPCR |
AURA - Positive Top-Line Results For 24 Week Data
On August 15, 2016, we announced positive top-line results from the AURA clinical trial in patients with active LN. The trial achieved its primary endpoint, demonstrating statistically significantly greater CR at 24 weeks (and confirmed at 26 weeks) in patients treated with 23.7 mg of voclosporin twice daily (p=0.045). This was the first global study of LN to meet its primary end point. Both treatment arms, 23.7 mg and 39.5 mg twice daily also showed a statistically significant improvement in the rate of achieving PR at 24 weeks (p=0.007; p=0.024). Each arm of the trial included the current standard of care of MMF as background therapy, and a forced steroid taper.
AURA Trial Design
The AURA clinical trial compared the efficacy of voclosporin added to current standard of care of MMF, also known as CellCept®, against standard of care with placebo in achieving CR in patients with active LN. It enrolled 265 patients at centers in 20 countries worldwide. On entry to the trial, patients were required to have a diagnosis of LN according to established diagnostic criteria (American College of Rheumatology) and clinical and biopsy features indicative of active LN.
Patients were randomized to one of two dosage groups of voclosporin (23.7 mg BID and 39.5 mg BID) or placebo, with all patients also receiving MMF and oral corticosteroids as background therapy. All patients had an initial IV dose of steroids (500-1000 mg) and then were started on 20-25 mg/daily, which was tapered down to a low dose of 5 mg daily by week 8 and 2.5 mg daily by week 16.
The primary endpoint was a measure of the number of patients who achieved CR at 24 weeks which had to be confirmed at 26 weeks. CR required the following four elements:
| |
• | protein/creatinine ratio of 0.5 mg/mg; |
| |
• | normal stable renal function (eGFR 60 mL/min/1.73m2 or no confirmed decrease from baseline in eGFR of 20%); |
| |
• | presence of sustained, low dose steroids (10mg/day of prednisone from week 16 - 24); and |
| |
• | no administration of rescue medications throughout the treatment period. |
Summary of Results
The groups were generally well-balanced for age, gender and race, however, when considered together, the proteinuria and eGFR data suggest that disease severity was greater for the low-dose voclosporin group.
Efficacy
| |
• | The primary endpoint of CR was met for the low-dose voclosporin group in the ITT analysis (p=0.045). 32.6% of patients on low dose achieved CR, compared to 27.3% on high dose and 19.3% in the control arm. |
| |
• | The odds ratio indicates that patients were twice as likely to achieve CR at 24 weeks compared to the control arm (OR=2.03). |
| |
• | The primary endpoint was re-analyzed using the 24-hour urine data in place of First Morning Void collections, confirming the finding that patients were twice as likely to achieve CR at 24 weeks compared to the control arm (p=0.047; OR=2.12). |
| |
• | Both voclosporin groups had a significantly faster time to CR (UPCR 0.5 mg/mg) than the control arm. Results of time to CR for co-variate analyses were broadly consistent with overall efficacy rates in those sub-groups. |
| |
• | The secondary endpoint of PR (50% reduction in UPCR over baseline with no administration of rescue medication throughout the treatment period) was met for both voclosporin groups in the ITT analysis with 69.7% of patients on low dose achieving PR (p=0.007) and 65.9% in the high dose group (p=0.024). 49.4% of patients in the control arm achieved PR. |
| |
• | Time to PR was similar (4 weeks) in the two voclosporin groups and was shorter than what was observed in the control group (6.6 weeks). |
Safety
| |
• | The overall rate of AEs was similar across all groups. |
| |
• | The overall rate of SAEs was higher in both voclosporin groups but the nature of SAEs is consistent with highly active LN. |
| |
• | The overall pattern of AEs and SAEs was consistent with that observed in other LN studies. |
| |
• | There were 13 deaths across the trial: two in the high-dose voclosporin arm; 10 in the low-dose voclosporin arm; and one in the control arm, with the majority of overall deaths (11/13) occurring in Asia. All deaths were assessed by the study investigator as being unrelated to study treatment. |
On September 29, 2016, we announced that in addition to voclosporin (23.7 mg BID) achieving its primary endpoint of CR at 24 weeks, both doses of voclosporin when added to the current standard of care of MMF and a forced oral corticosteroid taper have met all 24-week pre-specified secondary endpoints vs the control group. These pre-specified endpoints include: PR, which is measured by a 50% reduction in UPCR with no concomitant use of rescue medication; time to CR and PR; reduction in SLEDAI score; and reduction in UPCR over the 24-week treatment period.
|
| | | | | |
Pre-specified Secondary Endpoint | Control | | Low Dose VCS (23.7mg BID) | | High Dose VCS (39.5mg BID) |
Time to CR [median] | Not achieved | | 19.7 weeks p<.001 | | 23.4 weeks p=.001 |
PR (as measured by UPCR reduction of ≥ 50% from baseline) | 49% | | 70% p=.007 | | 66% p=.024 |
Time to PR [median] | 6.6 weeks | | 4.1 weeks p=.002 | | 4.4 weeks p=.003 |
SLEDAI Reduction | -4.5 | | -6.3 p=.003 | | -7.1 p=.003 |
Reduction in UPCR | -2.216 mg/mg | | -3.769 mg/mg p<.001 | | -2.792 mg/mg p=.006 |
| | | All p-values are vs control |
On September 30, 2016, we presented detailed results on the AURA clinical trial. These included a number of pre-specified subset and co-variate analyses and post-hoc analyses on the data, which show rapid proteinuria reduction and early remission. Based on recent literature suggesting that using a UPCR of .7mg/mg has better predictive power regarding long-term renal outcomes in LN patients, we performed a post hoc analysis applying this measure. In doing so, we saw both a greater treatment difference between the 23.7 mg BID voclosporin arm and the control arm, and better statistical power, which improves from a p-value of .045 to less than .01.
Based on these data we believe:
| |
• | voclosporin has shown statistically significant efficacy in multiple dimensions; |
| |
• | pre-specified and post-hoc analyses have provided valuable insight; |
| |
• | the LN Phase 3 clinical trial will be de-risked based upon the AURA results; and |
| |
• | biomarker data suggest significant effect on the underlying immunologic process of the disease. |
We also released detailed safety data for the trial including an in-depth mortality assessment. The safety and tolerability of voclosporin has been well-documented in numerous studies. In previous studies, over 2,000 patients have been treated with voclosporin across multiple indications with no unexpected SAEs. Clinical doses of voclosporin studies to date range from 13-70 mg BID.
In comparing four global LN trials, AURA, ALMS, Ocrelizumab and Abatacept, it is evident that the AURA clinical trial enrolled the most severe patients, as measured by proteinuria at baseline. The difference in UPCR and the eGFR in the low dose voclosporin arm at baseline indicates patients had more severe disease.
No new safety signals were observed with the use of voclosporin in LN patients and voclosporin was well- tolerated. The overall safety profile of voclosporin is consistent with other immunomodulators. The summary of AEs by SOC across arms in the trial is as follows:
|
| | | |
| Control | Voclosporin 23.7mg BID | Voclosporin 39.5 mg BID |
SOC | N=88 | N=89 | N=88 |
Any AE | 74 (84.1) | 81 (91.0) | 84 (95.5) |
Thirteen deaths have been reported in the AURA clinical trial which is a pattern that is consistent with other global active LN studies.
On November 15, 2016, at the American College of Rheumatology annual meeting, we presented speed of remission data from the AURA trial in a late-breaking abstract titled “Speed of Remission with the Use of Voclosporin, MMF and Low Dose Steroids: Results of a Global Lupus Nephritis Study.” The data presented are a post-hoc responder analysis (median time to CR for those who achieve CR), demonstrating 7.3 weeks to CR for voclosporin 23.7mg BID vs the control arm of 12 weeks.
On November 21, 2016, at the American Society of Nephrology Kidney Week 2016, we presented renal function data for the AURA trial in a late breaking session titled “High Impact Clinical Trials”. These data showed that in the voclosporin treatment arms, the renal function as measured by eGFR was stable and not significantly different from the control arm during the course of the trial. Mean blood pressure was slightly reduced and was similar between all treatment groups.
2016 Financings
June 2016 Private Placement
On June 22, 2016, we completed a private placement of 3 million units at US$2.36 per unit for aggregate gross proceeds of US$7.08 million. Each unit consisted of one Common Share and a 0.35 of one Common Share purchase warrant exercisable for a period of two years from the date of issuance at an exercise price of US$2.77.
July 2016 ATM
On July 22, 2016, we entered into a controlled equity offering sales agreement with Cantor Fitzgerald & Co. pursuant to which the Company was authorized to sell, from time to time, through at-the-market offerings with Cantor Fitzgerald & Co. acting as sales agent, such Common Shares as would have an aggregate offer price of up to US$10 million. We also filed a prospectus supplement with securities regulatory authorities in Canada in the provinces of British Columbia, Alberta and Ontario, and with the SEC, which supplemented our 2015 Base Shelf Prospectus and our Registration Statement. Sales in the July 2016 ATM were only conducted in the United States through NASDAQ at market prices. No sales were conducted in Canada or through the TSX.
As of October 3, 2016, sales pursuant to the July 2016 ATM were concluded. We issued 3.31 million Common Shares, receiving gross proceeds in the aggregate of US$8 million (US$6.14 million in the third quarter of 2016 and US$1.86 million subsequent to the quarter end), being the maximum value permissible in accordance with Canadian securities laws.
November 9, 2016 ATM
We entered into a controlled equity offering sales agreement with Cantor Fitzgerald & Co. dated November 9, 2016 relating to the sale of our Common Shares having an aggregate offering price of up to US$8.0 million. We also filed a prospectus supplement on November 9, 2016 with securities regulatory authorities in Canada in the provinces of British Columbia, Alberta and Ontario, and with the United States Securities and Exchange Commission, which supplemented our shelf prospectus. The prospectus supplement was amended, and an amended and restated prospectus supplement was filed on February 24, 2017 to update changes to certain information.
The sales under the November 2016 ATM were only conducted in the United States through NASDAQ at market prices. No sales were conducted in Canada or through the TSX.
As a result of completion of the March Offering, we determined that the November 2016 ATM facility was no longer required and as a result the sales agreement was terminated effective May 8, 2017.
As at December 31, 2016, we had issued 139,000 Common Shares and received gross proceeds of US$396,000. There were no sales under the November 2016 ATM in 2017.
December 2016 Public Offering
On December 28, 2016, we closed our US$28.75 million financing (including US$3.75 million pursuant to an exercise of the underwriters’ over-allotment option), for the sale of 12.78 million units at a price of US$2.25 per unit. Each unit consisted of one Common Share and one half of one Common Share purchase warrant. Each December 2016 Warrant entitles the holder to purchase one common share at the exercise
price of US$3.00 per common share for a period of five years after the closing of the offering. H.C. Wainwright & Co., LLC acted as sole book-running manager, and Cormark Securities Inc., acted as co-manager. The underwriters received a fee of 7.0% of the gross proceeds of the offering.
REGULATORY
Aurinia intends to submit for marketing approval in the United States, Europe and Japan based on the data from AURORA and the results of the AURA clinical trial.
REGULATORY REQUIREMENTS
The development, manufacturing and marketing of voclosporin is subject to regulations relating to the demonstration of safety and efficacy of the products as established by the government (or regulatory) authorities in those jurisdictions where this product is to be marketed. We would require regulatory approval in the United States, Europe and Japan where activities would be conducted by us or on our behalf. Depending upon the circumstances surrounding the clinical evaluation of the product candidate, the Company itself may undertake clinical trials, contract clinical trial activities to contract research organizations, or rely upon corporate partners for such development. We believe this approach will allow us to make cost effective developmental decisions in a timely fashion. We cannot predict or give any assurances as to whether regulatory approvals will be received or how long the process of seeking regulatory approvals will take.
Although only the jurisdictions of the United States, Europe and Japan are discussed in this section, we may also seek regulatory approval in other jurisdictions in the future and may initiate other clinical studies if and where appropriate.
United States
In the United States, all drugs are regulated under the Code of Federal Regulations and are enforced by the FDA. The regulations require that non-clinical and clinical studies be conducted to demonstrate the safety and effectiveness of products before marketing, and that the manufacturing be conducted according to certain GMP standards provided by the FDA.
Subsequent to the initial proof-of-concept and preliminary safety studies, the application submitted to the FDA prior to conducting human clinical trials of new drugs is referred to as an IND application. This application contains information related to the safety, efficacy and quality of the drug, and the FDA has 30 days in which to notify us if the application is unsatisfactory. If the application is deemed satisfactory, then we may proceed with the clinical trials. Before a clinical trial can commence at each participating clinical trial site, the site’s IRB/IEC must approve the clinical protocol and other related documents. The FDA or an IRB/IEC may place a hold on a clinical trial at any time.
After completing all required non-clinical and clinical trials, and prior to selling a novel drug in the United States, we must also comply with NDA procedures required by the FDA. The NDA procedure includes the submission of a package to demonstrate safety and efficacy of the novel drug and describe the manufacturing processes and controls. FDA approval of the submission, including agreement on labelling, is required prior to commercial sale or commercial distribution of the product in the United States. Pre- and/or post-approval inspections of manufacturing and testing facilities are necessary. The FDA may also conduct inspections of the clinical trial sites and the non-clinical laboratories conducting pivotal safety studies to ensure compliance with good clinical practice and good laboratory practice requirements. The FDA has the authority to impose certain post-approval requirements, such as post-market surveillance clinical trials. In addition, FDA approval can be withdrawn for failure to comply with any post-marketing requirements or for other reasons, such as the discovery of significant adverse effects.
Europe
In Europe, the evaluation of new products is coordinated by the EMA. The regulations are similar to those in the United States and require that non-clinical and clinical studies be conducted to demonstrate the safety and effectiveness of products before marketing, and that the manufacturing be conducted according to good manufacturing practice.
Subsequent to the initial proof-of-concept and preliminary safety studies, and prior to conducting human clinical trials, a CTA must be submitted to the competent authority in the country where the clinical trial will be conducted. This application contains similar information to United States IND. In Europe, the clinical trials are regulated by the European Clinical Trial Directive (2001/20/EC). As in the United States, before a clinical trial can commence at each participating clinical trial site, the site’s IRB/IEC must approve the clinical protocol and other related documents.
A major difference in Europe, when compared to the United States, is with the approval process. In Europe, there are different procedures that can be used to gain marketing authorization in the EU. The first procedure is referred to as the centralized procedure and requires that a single application be submitted to the EMA and, if approved, allows marketing in all countries of the EU. The centralized procedure is mandatory for certain types of medicines and optional for others. The second procedure is referred to as national authorization and has two options; the first is referred to as the mutual recognition procedure and requires that approval is gained from one member state, after which a request is made to the other member states to mutually recognize the approval, whilst the second is referred to as the decentralised procedure which requires a member state to act as the reference member state through a simultaneous application made to other member states.
Japan
Japan has a unique set of processes for the regulation of drugs. The PMDA is the main Regulatory Agency that oversees the review and approval of the drugs as per the regulatory prerequisites in Japan.
Japan’s regulatory system requires the IND Application documents to be prepared in the Common Technical Document (CTD) format. Subsequent to the application submission, PMDA evaluates the application with respect to the preclinical data, and protocols for clinical studies etc. It takes approximately 30 days for initial IND and 14 days for subsequent IND filings. Once queries have been answered by the applicant, PMDA completes its review and the IND application will be transferred to IRB for the review. IRB takes one to four weeks of time for the completion of the review. Once IRB provides a favorable response, IND application will be approved after which, clinical trials can be initiated on human subjects in Japan.
Once the applicant files the J-NDA, PMDA reviews the application and schedules a face-to-face meeting with the applicant during which queries from PMDA are discussed. Meanwhile, GMP investigation of manufacturing site will be carried out. After the face-to-face meeting, the PMDA reviewer prepares a Review Report. If there are any major issues, PMDA organizes the Expert Discussion, which involves a discussion between the PMDA reviewer and external expert on the proposed major issue(s). Subsequent to the discussions with the external expert, PMDA reviewer will prepare a summary of the main issues and discuss with the applicant in another face-to-face review meeting (can be held 2 times).
Following this review meeting, PMDA may again hold another Expert Discussion (if necessary) and prepares the Review Report for final approval within the Japanese government. The standard time for approval of a J-NDA is approximately 12 months.
BUSINESS MATTERS
DRUG DEVELOPMENT PROCESS
Clinical trials involve the administration of an investigational pharmaceutical product to individuals under the supervision of qualified medical investigators. Clinical studies are conducted in accordance with protocols that detail the objectives of a study, the parameters to be used to monitor safety, and the efficacy criteria to be evaluated. Each protocol is submitted to the appropriate regulatory body and to a relevant IRB/IEC prior to the commencement of each clinical trial. Clinical studies are typically conducted in three sequential phases which may overlap in time-frame.
In summary, the following steps must be completed prior to obtaining approval for marketing in the United States and Europe:
| |
1. | Nonclinical Animal Studies - These studies evaluate the safety and potential efficacy of a therapeutic product and form part of the application which must be reviewed by the appropriate regulatory authority prior to initiation of human clinical trials. |
| |
2. | Phase 1 Clinical Trials - These trials test the product in a small number of healthy volunteers to determine toxicity (safety), maximum dose tolerance, and pharmacokinetic properties. |
| |
3. | Phase 2 Clinical Trials - These trials are conducted in the intended patient population and include a larger number of subjects than in Phase 1. The primary goal is to determine the safety of a product in a larger number of patients and ultimately in the intended patient population. These trials may also provide early information on the potential effectiveness of a product. |
| |
4. | Phase 3 Clinical Trials - These trials are conducted in an expanded patient population at multiple sites to determine longer-term clinical safety and efficacy of the product. It is from the data generated in these trials that the benefit/risk relationship of a product is established, and the final drug labelling claims are defined. |
In the course of conducting clinical trials for a drug candidate, a company may conduct more than one trial of a particular phase in order to evaluate the drug against a variety of indications or in different patient populations. In such a case, industry practice is to differentiate these trials by way of designations such as “Phase 2a” or “Phase 2b”.
A key factor influencing the rate of progression of clinical trials is the rate at which patients can be recruited to participate in the research program. Patient recruitment is largely dependent upon the incidence and severity of the disease and the alternative treatments available.
Even after marketing approval for a drug has been obtained, further trials may be required (referred to as Phase 4 trials). Post-market trials may provide additional data on safety and efficacy necessary to gain approval for the use of the product as a treatment for clinical indications other than those for which the product was initially tested. These trials may also be used for marketing purposes.
MANUFACTURING, ENCAPSULATING AND PACKAGING OF VOCLOSPORIN
Drug supply costs are comprised of third party charges for manufacturing, encapsulating and packaging of voclosporin.
Voclosporin, requires a specialized manufacturing process. Lonza is currently our sole manufacturer of voclosporin and has manufactured the API for our clinical trials since 2004. Pricing for clinical supply is determined through negotiations between Lonza and the Company and is based on the size of specific API production runs and the cost of the raw materials used in the API manufacturing process. As at the date of this AIF, we have not experienced any difficulty in obtaining the raw materials required with respect to the manufacturing of voclosporin.
Lonza Manufacturing Collaboration Agreement
In November 2016, we entered into a long-term manufacturing collaboration and services agreement with Lonza for the manufacture of our API. This agreement follows a successful multi-year clinical manufacturing relationship where the Company and Lonza have been refining the process and analytical methods to produce clinical and commercial supplies of voclosporin. Under the terms of the agreement, Lonza has agreed to produce cGMP-grade voclosporin drug substance for use in our clinical trials and for future commercial use. The agreement also provides an option to have Lonza exclusively supply API for up to 20 years. Lonza is the sole supplier for manufacture of our API.
Encapsulating and Packaging of Voclosporin
We have contracted Catalent to encapsulate and package voclosporin for our LN and FSGS clinical studies. Catalent is currently the sole supplier for encapsulating and packaging our voclosporin clinical drug supply. Pricing for these services is determined by negotiations between Catalent and the Company and is based on the specific production run size.
It is our intention that Catalent will provide services with respect to encapsulating voclosporin required for our future commercial supply needs. We are currently in the process of determining our packaging supplier for our commercial supply requirements.
VOS
We have contracted Unither to manufacture VOS for our DES clinical studies. Sharp Clinical packages VOS for our clinical DES studies. Pricing for these services is determined by negotiations between Unither and Sharp Clinical, respectively, and the Company and is based on the specific production run size.
INTELLECTUAL PROPERTY RIGHTS
Patents and other proprietary rights are essential to our business. Our policy has been to file patent applications to protect technology, inventions, and improvements to our inventions that are considered important to the development of our business. We are pursuing certain avenues to expand the voclosporin intellectual property portfolio, including a use patent strategy (which involves potential development of use patents driven by AURA Phase 2b data) and a potential manufacturing patent and trade secret strategy.
The Company has an extensive granted patent portfolio related to cyclosporine analogs, including granted United States patents, covering voclosporin composition of matter, methods of use, formulations and synthesis. The corresponding Canadian, South African and Israeli patents are owned by Paladin Labs Inc. We anticipate that upon regulatory approval, patent protection for voclosporin will be extended in the United States (Patent Term Extension) and certain other major markets, including Europe and Japan, until at least October 2027 under the Hatch-Waxman Act in the United States and comparable patent extension laws in other countries (including the Supplementary Protection Certificate program in Europe). Opportunities may also be available to add an additional six months of exclusivity related to pediatric studies which are currently in the planning process. In addition to patent rights, we also expect to receive "new chemical entity" exclusivity for voclosporin in certain countries, which provides from five years in the United States and up to ten years in Europe.
Further, pursuant to a Notice of Allowance from the USPTO for claims directed at our novel voclosporin dosing protocol for LN as more fully discussed in the Recent Developments section of this AIF, after administrative processes are completed and fees are paid, we expect the issuance of a U.S. patent with a term extending to December 2037. If the FDA approves the use of voclosporin for LN and the label for such use follows the dosing protocol under the Notice of Allowance, the issuance of this patent will expand the scope of intellectual property protection for voclosporin, which already includes robust manufacturing, formulation, synthesis and composition of matter patents. We have also filed for protection of this subject matter under the PCT and have the option of applying for similar protection in the member countries thereof. This may lead to the granting of corresponding claims in the treaty countries which include all the major global pharmaceutical markets.
We have licensed the development and distribution rights to voclosporin for China, Hong Kong and Taiwan to 3SBio. This license is royalty bearing and we will also supply finished product to 3SBio on a cost-plus basis. We do not expect to receive any royalty revenue pursuant to this license in the foreseeable future.
We have patent protection for VOS as we own two granted United States patents and 14 patents in other jurisdictions related to ophthalmic formulations of calcineurin inhibitors or mTOR inhibitors, including voclosporin. We also have one granted United States patent and 10 patents in other jurisdictions related to topical drug delivery system for ophthalmic use. These patents expire between 2028 and 2031.
COMPETITIVE ENVIRONMENT
The pharmaceutical and biotechnology industries are characterized by rapidly evolving technology and intense competition. Many companies, including major pharmaceutical as well as specialized biotechnology companies, are engaged in activities focused on medical conditions that are the same as, or similar to, those targeted by us. Many of these companies have substantially greater financial and other resources, larger research and development staff, and more extensive marketing and manufacturing organization than we do. Many of these companies have significant experience in preclinical testing, human clinical trials, product manufacturing, marketing and distribution, and other regulatory approval procedures. In addition, colleges, universities, government agencies, and other public and private research organizations conduct research and may market commercial products on their own or through collaborative agreements. These institutions are becoming more active in seeking patent protection and licensing arrangements to collect royalties for use of technology that they have developed. These institutions also compete with us in recruiting and retaining highly qualified scientific personnel.
EMPLOYEES
|
| | | | | | | |
| As at December 31, 2018 | | As at December 31, 2017 | | As at December 31, 2016 |
Total Number of Employees | 39 | | 33 |
| | 20 |
|
As at December 31, 2018 we employed 39 employees, 34 of whom held advanced degrees in science and business, including two with a Ph.D. degree, two with a MD, one with a J.D. and seven with a Masters degree.
Of our total 39 employees as at December 31, 2018, 20 employees were engaged in, or directly support, clinical trial activities; and 19 employees were engaged in corporate, administration and business development activities.
Our employees are not governed by a collective agreement. We have not experienced a work stoppage and believe our employee relations are satisfactory given the current economic conditions.
FACILITIES
The Company entered into an agreement, effective June 1, 2014, to sublease 5,540 square feet of office and storage space at its head office location in Victoria, British Columbia for a term of five years. On December 6, 2018 the Company signed a commitment letter and entered into a new sublease on January 28, 2019 to rent 9,406 square feet of office and storage space at the existing location effective June 1, 2019. The new sublease is for a term of three years, however, the Company has the ability to cancel upon 12 months' notice. The estimated base rent plus operating costs on a monthly basis for the period from January 1, 2019 to May 31, 2019 is approximately US$11,000 per month increasing to approximately US$21,000 per month for the period of June 1, 2019 to May 31, 2022.
The Company entered into an agreement on November 14, 2014 to lease 1,247 square feet of office space for a term of two years commencing on January 1, 2015 for the Edmonton, Alberta registered office where the Company’s finance group is located. The lease was subsequently renewed until December 31, 2019 at a cost of approximately US$1,400 per month on the same terms as the original lease.
RISK FACTORS
Investing in our securities involves a high degree of risk. You should carefully consider the following risks in addition to the other information included in this AIF, our historical consolidated financial statements and related notes, before you decide to purchase our Common Shares. The risks and uncertainties described below are those that we currently believe may materially affect the Company and are set out in no particular order. Additional risks and uncertainties that we are unaware of or that we currently deem immaterial may also become important factors that materially and adversely affect our business, financial condition and results of operations. If any of the following events were to actually occur, our business, operating results or financial condition could be adversely affected in a material manner.
RISKS RELATING TO AURINIA'S BUSINESS
Clinical Trial Progress and Results - Heavy Dependence on Voclosporin
We have invested a significant portion of our time and financial resources in the development of voclosporin. We anticipate that our ability to generate revenues and meet expectations will depend primarily on the successful development, regulatory approval and commercialization of voclosporin.
The successful development and commercialization of voclosporin will depend on several factors, including the following:
| |
• | successful and timely completion of our clinical programs in LN and DES, including the AURORA trial which is anticipated to be completed in late 2019; |
| |
• | receipt of marketing approvals from the FDA and other regulatory authorities with a commercially viable label; |
| |
• | securing and maintaining sufficient expertise and resources to help in the continuing development and eventual commercialization of voclosporin; |
| |
• | maintaining suitable manufacturing and supply arrangements to ensure commercial quantities of the product through validated processes; |
| |
• | acceptance and adoption of the product by the medical community and third-party payers; and |
| |
• | our ability to raise future financial resources when required. Future additional sources of capital could include payments from equity financings, debt financings, potential new licensing partners, and/or the monetization of our intangible assets. |
It is possible that we may decide to discontinue the development of voclosporin at any time for commercial, scientific, or regulatory reasons. If voclosporin is developed, but not marketed, we will have invested significant resources and our future operating results and financial conditions would be significantly adversely affected. If we are not successful in commercializing voclosporin, or significantly delayed in doing so, our business will be materially harmed, and we may need to curtail or cease operations.
We may not be able to obtain required regulatory approvals for our product candidate and there is no assurance of successful development.
We have not completed the development of any therapeutic products and in particular, voclosporin, and therefore there can be no assurance that any product will be successfully developed. Voclosporin has not received regulatory approval for our commercial use and sale for any indication, in any jurisdiction. We cannot market a pharmaceutical product in any jurisdiction until it has completed thorough preclinical testing and clinical trials in addition to that jurisdiction’s extensive regulatory approval process. In general, significant research and development and clinical studies are required to demonstrate the safety and effectiveness of our product before submission of any regulatory applications. We may never obtain the required regulatory approvals for our product in any indication. Product candidates require significant additional research and development efforts, including clinical trials, prior to regulatory approval and potential commercialization, however, there can be no assurance that the results of all required clinical trials will demonstrate that these product candidates are safe and effective or, even if the results of all required clinical trials do demonstrate that these product candidates are safe and effective, or even if the results of the clinical trials are considered successful by us, that the regulatory authorities will not require us to conduct additional clinical trials before they will consider approving product candidates for commercial use. The FDA and other regulators have substantial discretion in the approval process.
Approval or consent by regulatory authorities to commence a clinical trial does not indicate that the device, drug, or treatment being studied can or will be approved. Of the large number of drugs in development, only a small percentage result in the submission of an application to the FDA and even fewer are approved for commercialization. The process of obtaining required approvals (such as, but not limited to, the approval of the FDA, the EMA, PMDA and Health Canada) is complex, expensive, time intensive, entails significant uncertainty and there can be no assurance that future products will be successfully developed, proven safe and effective in clinical trials or receive applicable regulatory approvals. Potential investors should be aware of the risks, problems, delays, expenses and difficulties which may be encountered by us in view of the extensive regulatory environment which controls our business. The regulatory review process typically varies in time, may take years to complete and approval is not guaranteed. Any approval might also contain significant limitations which may affect our ability to successfully develop its product candidate. Also, any regulatory approval once obtained, may be withdrawn. If regulatory approval is obtained in one jurisdiction, that does not necessarily mean that we will receive regulatory approval in all jurisdictions in which we may seek approval, or any regulatory approval obtained may not be as broad as what was obtained in other jurisdictions. However, the failure to obtain approval for our product candidate in one or more jurisdictions may negatively impact our ability to obtain approval in a different jurisdiction. If our development efforts for our product candidate are not successful or regulatory approval is not obtained in a timely fashion, on acceptable terms or at all, it will have a material adverse effect on the business, financial condition, and results of operations.
The results of our completed preclinical studies and clinical trials may not be indicative of future clinical trial results. A commitment of substantial resources to conduct time-consuming research, preclinical studies, and clinical trials will be required if we are to complete the development of our product.
There can be no assurance that unacceptable toxicities or adverse side effects will not occur at any time in the course of preclinical studies or human clinical trials or, if any products are successfully developed and approved for marketing, during commercial use of our product. The appearance of any such unacceptable toxicities or adverse side effects could interrupt, limit, delay, or abort the development of our product or, if previously approved, necessitate its withdrawal from the market. Furthermore, there can be no assurance that disease resistance or other unforeseen factors will not limit the effectiveness of our product. Any products resulting from our programs are not expected to be successfully developed or made commercially available in the near term and may not be successfully developed or made commercially available at all. Should our product prove to have insufficient benefit and/or have an unsafe profile, its development will likely be discontinued.
Our future performance will be impacted by a number of important factors, including, in the short-term, our ability to continue to generate cash flow from financings, and in the longer term, our ability to generate royalty or other revenues from licensed technology and bring new products to the market. Our future success will require efficacy and safety of our product and regulatory approval for the product. Future success of commercialization of any product is also dependent on our ability to obtain patents, enforce such patents, avoid patent infringement, and obtain patent extensions where applicable.
Government Regulation
The production and marketing of our product and our ongoing research and development activities are subject to regulation by numerous federal, provincial, state and local governmental authorities in the United States and any other countries where we may test or market our product. These laws require the approval of manufacturing facilities, including adhering to “good manufacturing” and/or “good laboratory” practices during production and storage, the controlled research and testing of products, governmental review and approval of submissions requiring manufacturing, pre-clinical and clinical data to establish the safety and efficacy of the product for each use sought in order to obtain marketing approval, and the control of marketing activities, including advertising and labeling. Failure to adhere to these requirements could invalidate our data.
If we secure regulatory approval, we would continue to be subject to extensive ongoing regulatory requirements. Manufacturing of approved drug products must comply with extensive regulations governing GMP. Manufacturers and their facilities are subject to continual review and periodic inspections. As we may be dependent on third parties for manufacturing, we will have limited ability to ensure that any entity manufacturing products on our behalf is doing so in compliance with applicable GMP requirements. Failure or delay by any manufacturer of our product to comply with GMP regulations or to satisfy regulatory inspections could have a material adverse effect on us, including potentially preventing us from being able to supply products for clinical trials or commercial sales. In addition, manufacturers may need to obtain approval from regulatory authorities for product, manufacturing, or labeling changes, which requires time and money to obtain and can cause delays in product availability. We are also required to comply with good distribution practices such as maintenance of storage and shipping conditions, as well as security of products, in order to ensure product quality determined by GMP is maintained throughout the distribution network. In addition, we are subject to regulations governing the import and export of our products.
Sales and marketing of pharmaceutical products are subject to extensive federal and provincial or state laws governing on-label and off-label advertising, scientific/educational grants, gifts, consulting and pricing and are also subject to consumer protection and unfair competition laws. Compliance with extensive regulatory and enforcement requirements requires training and monitoring of the sales force and other field personnel, which could impose a substantial cost on us. To the extent our product is marketed by collaborators, our ability to ensure their compliance with applicable regulations would be limited. In addition, we are subject to regulations governing the design, testing, control, manufacturing, distribution, labeling, quality assurance, packaging, storage, shipping, import and export of our product candidate.
There can be no assurance that we will be able to achieve or maintain regulatory compliance with respect to all or any part of our current or future products or that we will be able to timely and profitably produce our product while complying with applicable regulatory requirements. If we fail to maintain compliance, regulatory authorities may not allow the continuation of the drug development programs or require us to make substantial changes to the drug. Any such actions could have a material adverse effect on the business, financial condition, and results of operations.
Product Development Goals and Time Frames
We set goals for, and make public statements regarding, timing of the accomplishment of objectives material to our success, such as the commencement and completion of clinical trials, anticipated regulatory approval dates, and time of product launch. The actual timing of these events can vary dramatically due to factors such as delays or failures in clinical trials, the uncertainties inherent in the regulatory approval process, and delays in achieving product development, manufacturing, or marketing milestones necessary to commercialize our product. There can be no assurance that our clinical trials will be completed, that regulatory submissions will be made or receive regulatory approvals as planned, or that we will be able to adhere to the current schedule for the validation of manufacturing and launch of our product. If we fail to achieve one or more of these milestones as planned, the price of the Common Shares could decline.
We will have significant additional future capital needs in 2020 and beyond and there may be uncertainties as to our ability to raise additional funding in the future to meet these needs.
We will require significant additional capital resources to expand our business, in particular the further development of our product candidate, voclosporin, whether for LN or any other indication. Advancing our product candidate, marketing for our product, or acquisition and development of any new products or product candidates will require considerable resources and additional access to capital markets. In addition, our future cash requirements may vary materially from those now expected. For example, our future capital requirements may increase if:
| |
• | we experience unexpected or increased costs relating to preparing, filing, prosecuting, maintaining, defending and enforcing patent claims, or other lawsuits, brought by either us or our competition; |
| |
• | we experience scientific progress sooner than expected in our discovery, research and development projects, if we expand the magnitude and scope of these activities, or if we modify our focus as a result of our discoveries; |
| |
• | we are required to perform additional pre-clinical studies and clinical trials; or |
| |
• | we elect to develop, acquire or license new technologies, products or businesses. |
We could potentially seek additional funding through corporate collaborations and licensing arrangements or through public or private equity or debt financing. However, if capital market conditions in general, or with respect to life sciences companies such as ours, are unfavorable, our ability to obtain significant additional funding on acceptable terms, if at all, will be negatively affected. Additional financing that we may pursue may involve the sale of Common Shares which could result in significant dilution to our shareholders. If sufficient capital is not available, we may be required to delay our research and development projects, which could have a material adverse effect on our business, financial condition, prospects or results of operations.
Patents and Proprietary Technology
Patents and other proprietary rights are essential to our business. Our policy has been to file patent applications to protect technology, inventions, and improvements to our inventions that are considered important to the development of our business.
Our success will depend in part on our ability to obtain patents, defend patents, maintain trade secret protection and operate without infringing on the proprietary rights of others. Interpretation and evaluation of pharmaceutical patent claims present complex and often novel legal and factual questions. Accordingly, there is some question as to the extent to which biopharmaceutical discoveries and related products and processes can be effectively protected by patents. As a result, there can be no assurance that:
| |
• | patent applications will result in the issuance of patents; |
| |
• | additional proprietary products developed will be patentable; |
| |
• | patents issued will provide adequate protection or any competitive advantages; |
| |
• | patents issued will not be successfully challenged by third parties; |
| |
• | our products do not infringe the patents or intellectual property of others; or |
| |
• | that we will be able to obtain any extensions of the patent term. |
A number of pharmaceutical, biotechnology, medical device companies and research and academic institutions have developed technologies, filed patent applications or received patents on various technologies that may be related to our business. Some of these technologies, applications or patents may conflict with or adversely affect our technologies or intellectual property rights. Any conflicts with the intellectual property of others could limit the scope of the patents, if any, that we may be able to obtain or result in the denial of patent applications altogether.
Further, there may be uncertainty as to whether we may be able to successfully defend any challenge to our patent portfolio. Moreover, we may have to participate in interference proceedings in the various jurisdictions around the world. An unfavorable outcome in an interference or opposition proceeding or a conflict with the intellectual property of others could preclude us or our collaborators or licensees from making, using or selling products using the technology, or require us to obtain license rights from third parties. It is not known whether any prevailing party would offer a license on commercially acceptable terms, if at all. Further, any such license could require the expenditure of substantial time and resources and could harm our business. If such licenses are not available, we could encounter delays or prohibition of the development or introduction of our product.
We received a Notice of Allowance from the USPTO for claims directed at our novel voclosporin dosing protocol for LN (U.S. patent application 15/835,219, entitled "PROTOCOL FOR TREATMENT OF LUPUS NEPHRITIS”) which concluded a substantive examination of the patent application at the USPTO, and after administrative processes are completed and fees are paid, is expected to result in the issuance of a U.S. patent with a term extending to December 2037. For this patent to be useful, it will require that the FDA approve the use of voclosporin for LN and that the label for such use will follows the dosing protocol under the Notice of Allowance claims.
Clinical trials for our product candidate are expensive and time-consuming, and their outcome is uncertain.
Before we can obtain regulatory approval for the commercial sale of any product candidate currently under development, we are required to complete extensive clinical trials to demonstrate its safety and efficacy. Clinical trials are very expensive and difficult to design and implement. The clinical trial process is also time-consuming. If we find a collaboration partner for the development of voclosporin (whether for LN, DES or any other indication), the clinical trials are expected to continue for several years, although costs associated with voclosporin may well be shared with our collaboration partner. The timing of the commencement, continuation and completion of clinical trials may be subject to significant delays relating to various causes, including:
| |
• | our inability to find collaboration partners, if needed; |
| |
• | our inability to manufacture or obtain sufficient quantities of materials for use in clinical trials; |
| |
• | delays in obtaining regulatory approvals to commence a study, or government intervention to suspend or terminate a study; |
| |
• | delays, suspension, or termination of the clinical trials imposed by the IRB/IEC responsible for overseeing the study to protect research subjects at a particular study site; |
| |
• | delays in identifying and reaching agreement on acceptable terms with prospective clinical trial sites; |
| |
• | slower than expected rates of patient recruitment and enrollment; |
| |
• | uncertain dosing issues; |
| |
• | inability or unwillingness of medical investigators to follow our clinical protocols; |
| |
• | variability in the number and types of subjects available for each study and resulting difficulties in identifying and enrolling subjects who meet trial eligibility criteria; |
| |
• | scheduling conflicts with participating clinicians and clinical institutions; |
| |
• | difficulty in maintaining contact with subjects after treatment, which results in incomplete data; |
| |
• | unforeseen safety issues or side effects; |
| |
• | lack of efficacy during the clinical trials; |
| |
• | our reliance on clinical research organizations to conduct clinical trials, which may not conduct those trials with good clinical or laboratory practices; or |
| |
• | other regulatory delays. |
The results of pre-clinical studies and initial clinical trials are not necessarily predictive of future results, and our current product candidate may not have favourable results in later trials or in the commercial setting.
Success in pre-clinical or animal studies and early clinical trials neither ensure that later large-scale efficacy trials will be successful, nor does it predict final results. Pre-clinical tests and Phase 1 and Phase 2 clinical trials are primarily designed to test safety, to study pharmacokinetics and pharmacodynamics and to understand the side effects of product candidates at various doses and schedules. Favourable results in early trials may not be repeated in later trials.
A number of companies in the life sciences industry have suffered significant setbacks in advanced clinical trials, even after positive results in earlier trials. Clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals. Negative or inconclusive results or adverse medical events during a clinical trial could cause a clinical trial to be delayed, repeated or terminated. In addition, failure to construct appropriate clinical trial protocols could result in the test or control group experiencing a disproportionate number of AEs and could cause a clinical trial to be repeated or terminated. Pre-clinical data and the clinical results we have obtained for voclosporin (for LN or any other indication) may not predict results from studies in larger numbers of subjects drawn from more diverse populations or in a commercial setting, and also may not predict the ability of our product to achieve its intended goals, or to do so safely.
We will be required to demonstrate in Phase 3 clinical trials that voclosporin is safe and effective for use in a diverse population before we can seek regulatory approvals for its commercial sale. There is typically an extremely high rate of attrition from the failure of product candidates proceeding through clinical and post-approval trials. If voclosporin fails to demonstrate sufficient safety and efficacy in ongoing or future clinical trials, we could experience potentially significant delays in, or be required to abandon development of, our product candidate currently under development.
Our industry is subject to health and safety risks.
While we take substantial precautions such as laboratory and clinical testing, toxicology studies, quality control and assurance testing and controlled production methods, the health and safety risks associated with producing a product for human ingestion cannot be eliminated. Products produced by us may be found to be, or to contain substances that are harmful to the health of our patients and customers and which, in extreme cases, may cause serious health conditions or death. This sort of finding may expose us to substantial risk of litigation and liability.
Further, we would be forced to discontinue production of our product, which would harm our profitability. We maintain product liability insurance coverage; however, there is no guarantee that our current coverage will be sufficient or that we can secure insurance coverage in the future at commercially viable rates or with the appropriate limits.
Our product may not achieve or maintain expected levels of market acceptance, which could have a material adverse effect on our business, financial condition and results of operations and could cause the market value of our Securities to decline.
Even if we are able to obtain regulatory approvals for our product, the success of the product is dependent upon achieving and maintaining market acceptance. New product candidates that appear promising in development may fail to reach the market or may have only limited or no commercial success. Levels of market acceptance for our product could be impacted by several factors, many of which are not within our control, including but not limited to:
| |
• | safety, efficacy, convenience and cost-effectiveness of our product compared to products of our competitors; |
| |
• | scope of approved uses and marketing approval; |
| |
• | timing of market approvals and market entry; |
| |
• | difficulty in, or excessive costs to, manufacture; |
| |
• | infringement or alleged infringement of the patents or intellectual property rights of others; |
| |
• | availability of alternative products from our competitors; |
| |
• | formulary placement and status; |
| |
• | acceptance of the price of our product; and |
| |
• | ability to market our product effectively at the retail level. |
In addition, by the time any products are ready to be commercialized, what we believe to be the market for these products may have changed. Our estimates of the number of patients who have received or might have been candidates to use a specific product may not accurately reflect the true market or market prices for such products or the extent to which such products, if successfully developed, will actually be used by patients. Our failure to successfully introduce and market our products would have a material adverse effect on our business, financial condition, and results of operations.
We are dependent upon key personnel to achieve our business objectives.
Our ability to retain key personnel and attract other qualified individuals is critical to our success. As a technology-driven company, intellectual input from key management and personnel is critical to achieve our business objectives. The loss of the services of key individuals might significantly delay or prevent achievement of our business objectives. In addition, because of a relative scarcity of individuals with experience and the high degree of education and scientific achievement required for our business, competition among life sciences companies for qualified employees is intense and, as a result, we may not be able to attract and retain such individuals on acceptable terms, or at all. In addition, because we do not maintain “key person” life insurance on any of our officers, employees, or consultants, any delay in replacing such persons, or an inability to replace them with persons of similar expertise, would have a material adverse effect on our business, financial condition, and results of operations.
We also have relationships with scientific collaborators at academic and other institutions, some of whom conduct research at our request or assist us in formulating our research and development strategies. These scientific collaborators are not our employees and may have commitments to, or consulting or advisory contracts with, other entities that may limit their availability to us. In addition, even though our collaborators are required to sign confidentiality agreements prior to working with us, they may have arrangements with other companies to assist such other companies in developing technologies that may prove competitive to us.
Incentive provisions for our key executives include the granting of stock options that vest over time, designed to encourage such individuals to stay with us. However, a low share price, whether as a result of disappointing progress in our development programs or as a result of market conditions generally, could render such agreements of little value to our key executives. In such event, our key executives could be susceptible to being hired away by our competitors who could offer a better compensation package. If we are unable to attract and retain key personnel, our business, financial conditions and results of operations may be adversely affected.
We are exposed to risks relating to the write-down of intangible assets, which comprises a significant portion of our total assets.
A significant amount of our total assets relate to our intellectual property. As of December 31, 2018, the carrying value of our intangible assets was approximately US$12.62 million. In accordance with IFRS, we are required to review the carrying value of its intangible assets for impairment periodically or when certain triggers occur. Such impairment will result in a write-down of the intangible asset and the write-down is charged to income during the period in which the impairment occurs. The write-down of any intangible assets could have a material adverse effect on our business, financial condition, and results of operations.
If we were to lose our foreign private issuer status under U.S. federal securities laws, we would likely incur additional expenses associated with compliance with the U.S. securities laws applicable to U.S. domestic issuers.
As a foreign private issuer, as defined in Rule 3b-4 under the Exchange Act, we are exempt from certain of the provisions of the U.S. federal securities laws. For example, the U.S. proxy rules and the Section 16 reporting and “short swing” profit rules do not apply to foreign private issuers. However, if we were to lose our status as a foreign private issuer, these regulations would immediately apply and we would also be required to commence reporting on forms required of U.S. companies, such as Forms 10-K, 10-Q and 8-K, rather than the forms currently available to us, such as Forms 40-F and 6-K. Compliance with these additional disclosure and timing requirements under these securities laws would likely result in increased expenses and would require our management to devote substantial time and resources to comply with new regulatory requirements. Further, to the extent that we were to offer or sell our Securities outside of the United States, we would have to comply with the more restrictive Regulation S requirements that apply to U.S. companies, and we would no longer be able to utilize the multijurisdictional disclosure system forms for registered offerings by Canadian companies in the United States, which could limit our ability to access the capital markets in the future.
Legislative actions, potential new accounting pronouncements, and higher insurance costs are likely to impact our future financial position or results of operations.
Future changes in financial accounting standards may cause adverse, unexpected revenue fluctuations and affect our financial position or results of operations. New pronouncements and varying interpretations of pronouncements have occurred with greater frequency and are expected to occur in the future. Compliance with changing regulations of corporate governance and public disclosure may result in additional expenses. All of these uncertainties are leading generally toward increasing insurance costs, which may adversely affect our business, results of operations and our ability to purchase any such insurance, at acceptable rates or at all, in the future.
We rely on third parties for the supply and manufacture of voclosporin, which can be unpredictable in terms of quality, cost, timing and availability.
Our drug, voclosporin, requires a specialized manufacturing process. Lonza is currently the sole source manufacturer of voclosporin.
We have contracted Catalent to encapsulate and package voclosporin for our AURORA clinical trial program. Catalent is currently the sole supplier for encapsulating and packaging our clinical drug supply.
It is our intention that Catalent will provide services with respect to encapsulating the voclosporin required for future clinical and commercial supply needs, while the provider of packaging services for commercial supply is yet to be determined.
We have contracted Unither to manufacture VOS for our DES clinical studies, and we have contracted Sharp Clinical to package VOS for our clinical DES studies.
The FDA and other regulatory authorities require that drugs be manufactured in accordance with the current GMP regulations, as established from time to time. Accordingly, in the event we receive marketing approvals for voclosporin, it may need to rely on a limited number of third parties to manufacture and formulate voclosporin. We may not be able to arrange for our product to be manufactured on reasonable terms or in sufficient quantities.
Manufacturers of pharmaceutical products often encounter difficulties in production, especially in scaling up initial production. These problems include difficulties with production costs and yields, stability, quality control and assurance, and shortages of qualified personnel, as well as compliance with strictly enforced federal, provincial and foreign regulations. We rely on a limited number of third parties to manufacture and supply raw materials for our product. The third parties we choose to manufacture and supply raw materials for our product are not under our control and may not perform as agreed or may terminate their agreements with us, and we may not be able to find other third parties to manufacture and supply raw materials on commercially reasonable terms, or at all. If either of these events were to occur, our operating results and financial condition would be adversely affected.
In addition, drug and chemical manufacturers are subject to various regulatory inspections, including those conducted by the FDA, to ensure strict compliance with GMP and other government regulations. While we are obligated to audit the performance of our third-party contractors, we do not have complete control over their compliance. We could be adversely impacted if our third-party manufacturers do not comply with these standards and regulations. For non-compliance, the regulatory authority may levy penalties and sanctions, including fines, injunctions, civil penalties, failure of the government to grant review of submissions or market approval of drugs, or cause delays, suspension or withdrawal of approvals, product seizures or recalls, operating restrictions, facility closures and criminal prosecutions. Any of this will have a material adverse impact on our business, financial condition, and results of operations.
Anticipated revenues may be derived from Licensing Activities.
We anticipate that our revenues in the future may be derived from products licensed to pharmaceutical and biotechnology companies. Accordingly, these revenues will depend, in large part, upon the success of these companies, and our operating results may fluctuate substantially due to reductions and delays in their research, development and marketing expenditures. These reductions and delays may result from factors that are not within our control, including:
| |
• | changes in economic conditions; |
| |
• | changes in the regulatory environment, including governmental pricing controls affecting health care and health care providers; |
| |
• | other factors affecting research and development spending. |
Lack of Operating Profits
We have incurred losses and anticipate that our losses will increase as we continue the development of voclosporin and clinical trials and seek regulatory approval for the sale of our therapeutic product. There can be no assurance that we will have earnings or positive cash flow in the future.
As at December 31, 2018, we had an accumulated deficit of US$415.96 million. The net operating losses over the near-term and the next several years are expected to continue as a result of initiating new clinical trials and activities necessary to support regulatory approval and commercialization of our product. There can be no assurance that we will be able to generate sufficient product revenue to become profitable at all or on a sustained basis. We expect to have quarter-to-quarter fluctuations in expenses, some of which could be significant, due to research, development, and clinical trial activities, as well as regulatory and commercialization activities.
Negative Cash Flow
We had negative operating cash flow for the financial year ended December 31, 2018. We anticipate that we will continue to have negative cash flow as we continue our development of voclosporin. To the extent that we have negative operating cash flow in future periods, we will likely need to allocate a portion of our cash reserves to fund such negative cash flow. We may also be required to raise additional funds through the issuance of equity or debt securities. There can be no assurance that we will be able to generate a positive cash flow from our operations, that additional capital or other types of financing will be available when needed or that these financings will be on terms favourable or acceptable to us.
We may not realize the anticipated benefits of acquisitions or product licenses and integration of these acquisitions and any products acquired or licensed may disrupt our business and management.
As part of our business strategy, we may acquire additional companies, products or technologies principally related to, or complementary to, our current operations. At any given time, we may be evaluating new acquisitions of companies, products or technologies or may be exploring new licensing opportunities, and may have entered into confidentiality agreements, non-binding letters of intent or may be in the process of conducting due diligence with respect to such opportunities. Any such acquisitions will be accompanied by certain risks including, but not limited to:
| |
• | exposure to unknown liabilities of acquired companies and the unknown issues with any associated technologies or research; |
| |
• | higher than anticipated acquisition costs and expenses; |
| |
• | the difficulty and expense of integrating operations, systems, and personnel of acquired companies; |
| |
• | disruption of our ongoing business; |
| |
• | inability to retain key customers, distributors, vendors and other business partners of the acquired company; |
| |
• | diversion of management’s time and attention; and |
| |
• | possible dilution to shareholders. |
We may not be able to successfully overcome these risks and other problems associated with acquisitions and this may adversely affect our business, financial condition or results of operations.
Our business depends heavily on the use of information technologies.
Several key areas of our business depend on the use of information technologies, including production, manufacturing and logistics, as well as clinical and regulatory matters. Despite our best efforts to prevent such behavior, third parties may nonetheless attempt to hack into our systems and obtain data relating to our pre-clinical studies, clinical trials, patients using our product or our proprietary information on voclosporin. If we fail to maintain or protect our information systems and data integrity effectively, we could have problems in determining product cost estimates and establishing appropriate pricing, have difficulty preventing, detecting, and controlling fraud, have disputes with physicians, and other health care professionals, have regulatory sanctions or penalties imposed, have increases in operating expenses, incur expenses or lose revenues as a result of a data privacy breach, or suffer other adverse consequences. While we have invested in the protection of data and information technology, there can be no assurance that our efforts or those of our third-party collaborators, if any, or manufacturers, to implement adequate security and quality measures for data processing would be sufficient to protect against data deterioration or loss in the event of a system malfunction, or to prevent data from being stolen or corrupted in the event of a security breach. Any such loss or breach could have a material adverse effect on our business, operating results and financial condition.
Competition and Technological Change
The industry in which we operate is highly competitive and we have numerous domestic and foreign competitors, including major pharmaceutical and chemical companies, specialized biotechnology companies, universities, academic institutions, government agencies, public and private research organizations and large, fully-integrated pharmaceutical companies which have extensive resources and experience in research and development, process development, clinical evaluation, manufacturing, regulatory affairs, distribution and marketing. Many of our potential competitors possess substantially greater research and development skills, financial, technical and marketing expertise and human resources
than we do, and may be better equipped to develop, manufacture and market products. There is a risk that new products and technologies may be developed which may be more effective or commercially viable than the product being developed or marketed by us, thus making our product non-competitive or obsolete. There may also be market resistance to the acceptance of our new product in any indication and a risk that the product, even though clinically effective, is not economically viable in the commercial production stage.
Reliance on Partners
Our strategy and success for the research, development, and commercialization of voclosporin in China is dependent upon the activities of third parties with rights to voclosporin in those jurisdictions. The amount and timing of resources such third parties will devote to these activities may not be within our control. There can be no assurance that those third parties will perform as expected.
The license, research and development agreements with the third parties referenced above include indemnification and obligation provisions that are customary in the industry. These guarantees generally require us to compensate the other party for certain damages and costs incurred as a result of third party claims or damages arising from these transactions. These provisions may survive termination of the underlying agreement. The nature of the potential obligations prevents us from making a reasonable estimate of the maximum potential amount we could be required to pay.
Reliance on Other Third Parties
We depend on third parties for the sourcing of components or for the product itself. Furthermore, as with other pharmaceutical companies, we rely on medical institutions for testing and clinically validating our prospective product. We do not anticipate any difficulties in obtaining required components or products or any difficulties in the validation and clinical testing of our product but there is no guarantee that they will be obtained.
We currently rely on CROs for the conduct of our clinical trials. These CROs operate in accordance with good clinical management practices mandated by the regulatory authorities and are subject to regular audits by regulatory authorities and by us.
We also have arrangements for the encapsulation, packaging and labeling of voclosporin through third party suppliers. Contract manufacturers must operate in compliance with regulatory requirements. Failure to do so could result in, among other things, the disruption of product supplies.
Marketing and Distribution
We have limited experience in the sales, marketing, and distribution of pharmaceutical products. There can be no assurance that we will be able to establish sales, marketing, and distribution capabilities or make arrangements through collaborations, licensees, or others to perform such activities, or that such efforts would be successful. If we decide to market our product directly, we must either acquire or internally develop a marketing and sales force with technical expertise and provide supporting distribution capabilities. The acquisition or development of a sales and distribution infrastructure would require substantial resources, which may divert the attention of management and key personnel and have a negative impact on product development. If we contract with third parties for the sales and marketing of our product, our revenue will be dependent on the efforts of these third parties, whose efforts may not be successful. If we fail to establish successful marketing and sales capabilities or to make arrangements with third parties, the business, financial condition and results of operations will be materially adversely affected.
Health Care Reimbursement
In both domestic and foreign markets, sales of our product, if any, will be dependent in part on the availability of reimbursement from third party payors, such as government and private commercial insurance plans. Third party payors are increasingly challenging the prices charged for medical products and services. There can be no assurance that our product will be considered cost effective by these third-party payors, that reimbursement will be available or if available that the payor’s reimbursement policies will not adversely affect our ability to sell our product on a profitable basis.
Unauthorized Disclosure of Confidential Information
There may be an unauthorized disclosure of the significant amount of confidential information under our control. We maintain and manage confidential information relating to our technology, research and development, production, marketing and business operations and those of our collaborators, in various forms. Although we have implemented controls to protect the confidentiality of such information, there can be no assurance that such controls will be effective. Unauthorized disclosures of such information could subject us to complaints or lawsuits for damages, in Canada or other jurisdictions, or could otherwise have a negative impact on our business, financial condition, results of operations, reputation and credibility.
Use of Hazardous Materials
Drug manufacturing processes involve the controlled use of hazardous materials. We and our third-party manufacturing contractors are subject to regulations governing the use, manufacture, storage, handling and disposal of such materials and certain waste products. Although we believe that our third-party manufacturers have the required safety procedures for handling and disposing of such materials and comply with the standards prescribed by such laws and regulations, the risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of such an accident, we could be held liable for any damages that result and such liability could exceed our resources.
Liability and Insurance
The testing, marketing and sale of human pharmaceutical products involves unavoidable risks. If we succeed in developing new pharmaceutical products, the sale of such products may expose us to potential liability resulting from the use of such products. Such liability might result from claims made directly by consumers or by regulatory agencies, pharmaceutical companies or others. The obligation to pay any product liability claim in excess of whatever insurance we are able to acquire, or the recall of any of our products, could have a material adverse effect on our business, financial condition and future prospects.
We entered into indemnification agreements with our officers and directors. The maximum potential amount of future payments required under these indemnification agreements is unlimited. However, we currently maintain director and officer liability insurance coverage of US$35 million to reduce our exposure.
Financial instruments and Risks
We are exposed to credit risks and market risks related to changes in interest rates and foreign currency exchange, each of which could affect the value of our current assets and liabilities. We invest our cash reserves in U.S. dollar denominated, fixed rate, highly liquid and highly rated financial instruments such as treasury notes, banker acceptances, bank bonds, and term deposits. We do not believe that the results of operations or cash flows would be affected to any significant degree by a sudden change in market interest rates relative to our investment portfolio, due to the short-term nature of the investments and our current ability to hold these investments to maturity.
We are exposed to financial risk related to the fluctuation of foreign currency exchange rates which could have a material effect on our future operating results or cash flows. Foreign currency risk is the risk that variations in exchange rates between the United States dollar and foreign currencies, primarily with the Canadian dollar, will affect our operating and financial results. We hold our cash reserves in US dollars and the majority of our expenses, including clinical trial costs are also denominated in US dollars, which mitigates the risk of material foreign exchange fluctuations.
RISKS RELATED TO OUR SECURITIES
There is no assurance of a sufficient liquid trading market for our Common Shares in the future.
Our shareholders may be unable to sell significant quantities of Common Shares into the public trading markets without a significant reduction in the price of their Common Shares, or at all. There can be no assurance that there will be sufficient liquidity of our Common Shares on the trading market, and that we will continue to be listed on the TSX or the NASDAQ or achieve listing on any other public listing exchange.
Raising additional capital may cause dilution to our shareholders, restrict our operations or require us to relinquish rights to our technologies or drug candidate.
In order to meet our future financing needs, we may issue a significant amount of additional Common Shares, Warrants, subscription receipts, debt securities, Units, or other equity or debt securities. The precise terms of any future financing will be determined by us and potential investors and such future financings may significantly dilute our shareholders’ percentage ownership. Additionally, if we raise additional funds through collaborations, strategic alliances or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or drug candidate or grant licenses on terms that may not be favourable to us and/or that may reduce the value of the Common Shares.
Volatility of Share Price
The market prices for the securities of biotechnology companies, including ours, have historically been volatile. The market has from time to time experienced significant price and volume fluctuations that are unrelated to the operating performance of any particular company.
The trading price of the Common Shares could continue to be subject to wide fluctuations in price in response to various factors, many of which are beyond our control, including the results and adequacy of our preclinical studies and clinical trials, as well as those of our collaborators, or our competitors; other evidence of the safety or effectiveness of our products or those of our competitors; announcements of technological innovations or new products by us or our competitors; governmental regulatory actions; developments with collaborators; developments (including litigation) concerning our patent or other proprietary rights of competitors; concern as to the safety of our products; period-to-period fluctuations in operating results; changes in estimates of our performance by securities analysts; market conditions for biotechnology stocks in general; and other factors not within our control could have a significant adverse impact on the market price of the Common Shares, regardless of our operating performance. In the past, following periods of volatility in the market price of a company’s securities, securities class action litigation has often been instituted. A class action suit against us could result in substantial costs, potential liabilities and the diversion of management’s attention and resources.
There is no guarantee that an active trading market for the Common Shares will be maintained on either the TSX or NASDAQ. Investors may not be able to sell their Common Shares quickly or at the latest market price if the trading in the Common Shares is not active.
We expect to issue Common Shares in the future. Future issuances of Common Shares, or the perception that such issuances are likely to occur, could affect the prevailing trading prices of the Common Shares. In addition, the existence of Warrants or debt securities with conversion features may encourage short selling by market participants.
Sales of Common Shares could cause a decline in the market price of the Common Shares. One of our major shareholders (ILJIN and its affiliates) owns an aggregate of approximately 14.97% of our outstanding Common Shares as at March 15, 2019. Any sales of Common Shares by these shareholders or other existing shareholders or holders of options may have an adverse effect on our ability to raise capital and may adversely affect the market price of the Common Shares.
Future issuances of equity securities by us or sales by our existing shareholders may cause the price of the Common Shares to fall.
The market price of the Common Shares could decline as a result of issuances of Securities or sales by our existing shareholders in the market, or the perception that these sales could occur. Sales of Common Shares by shareholders might also make it more difficult for us to sell Common Shares at a time and price that we deem appropriate. With an additional sale or issuance of Common Shares, investors will suffer dilution of their voting power and may experience dilution in earnings per share.
We may have broad discretion in the use of the net proceeds of an offering of the Securities and may not use them to effectively manage our business.
We may need to exercise broad discretion over the use of the net proceeds from a future offering of Common Shares. Because of the number and variability of factors that will determine our use of such proceeds, our ultimate use might vary substantially from our planned use. Investors may not agree with how we allocate or spend the proceeds from an offering of Common Shares. We may pursue acquisitions, collaborations or clinical trials that do not result in an increase in the market value of the Common Shares and may increase our losses.
We do not intend to pay dividends in the foreseeable future.
We have never declared or paid any dividends on the Common Shares. We intend, for the foreseeable future, to retain our future earnings, if any, to finance our commercial activities and further research and the expansion of our business. As a result, the return on an investment in Common Shares will likely depend upon any future appreciation in value, if any, and on a shareholder’s ability to sell Common Shares. The payment of future dividends, if any, will be reviewed periodically by our Board and will depend upon, among other things, conditions then existing including earnings, financial conditions, cash on hand, financial requirements to fund our commercial activities, development and growth, and other factors that our Board may consider appropriate in the circumstances.
We may be a PFIC for U.S. tax purposes, which may result in adverse tax consequences for U.S. investors.
If we are characterized as a PFIC, there may be adverse tax consequences for U.S. investors. Generally, if for any taxable year 75% or more of our gross income is passive income, or at least 50% of the average quarterly value of our assets are held for the production of, or produce, passive income, we would be characterized as a PFIC for U.S. federal income tax purposes. Based on the nature of our income and the value and composition of our assets, we do not believe we were a PFIC during 2018. While we also do not believe we will be a PFIC for the current taxable year, because PFIC status is determined on an annual basis and generally cannot be determined until the end of the taxable year, there can be no assurance that we will not be a PFIC for the current or future taxable years. If we are characterized as a PFIC, our shareholders who are U.S. holders may suffer adverse tax consequences, including the treatment of gains realized on the sale of our ordinary shares as ordinary income, rather than as capital gain, the loss of the preferential rate applicable to dividends received on our ordinary shares by individuals who are U.S. holders, and the addition of interest charges to the tax on such gains and certain distributions. A U.S. shareholder of a PFIC generally may mitigate these adverse U.S. federal income tax consequences by making a “qualified electing fund” election, or, to a lesser extent, a “mark to market” election.
You may be unable to enforce actions against us, or certain of our directors and officers under U.S. federal securities laws.
As a corporation organized under the laws of Alberta, Canada, it may be difficult to bring actions under U.S federal securities law against us. Most of our directors and officers reside principally in Canada or outside of the United States. Because all or a substantial portion of our assets and the assets of these persons are located outside of the United States, it may not be possible for investors to effect service of process within the United States upon us or those persons. Furthermore, it may not be possible for investors to enforce against us or those persons in the United States, judgments obtained in U.S. courts based upon the civil liability provisions of the U.S. federal securities laws or other laws of the United States. There is doubt as to the enforceability, in original actions in Canadian courts, of liabilities based upon U.S. federal securities laws and as to the enforceability in Canadian courts of judgments of U.S. courts obtained in actions based upon the civil liability provisions of the U.S. federal securities laws. Therefore, it may not be possible to enforce those actions against us or certain of our directors and officers.
Adverse capital market conditions could affect out liquidity.
Adverse capital market conditions could affect our ability to meet our liquidity needs, as well as our access to capital and cost of capital. We need additional funding to continue development of our internal pipeline and collaborations in the future. Our results of operations, financial condition, cash flows and capital position could be materially affected by disruptions in the capital markets.
DIVIDEND POLICY
We have not paid dividends on our outstanding Common Shares in the past and have no established dividend policy for our Common Shares. We plan to use future earnings, if any, to finance further research and development and the expansion of our business and do not anticipate paying out dividends on our Common Shares in the foreseeable future. The payment of future dividends, if any, will be reviewed periodically by our Board and will depend upon, among other things, conditions then existing including earnings, financial conditions, cash on hand, financial
requirements to fund our commercial activities, development and growth, and other factors that our Board may consider appropriate in the circumstances.
CAPITAL STRUCTURE
The Company’s authorized share capital consists of an unlimited number of Common Shares, all without nominal or par value.
The holders of Common Shares are entitled to receive notice of and attend all meetings of shareholders, with each Common Share held entitling the holder to vote on any resolution to be passed at such shareholder meetings. The holders of Common Shares are entitled to dividends if, as and when declared by the Board. The Common Shares are entitled upon liquidation, dissolution or winding up of Aurinia, to receive the remaining assets of Aurinia available for distribution to shareholders. There are no pre-emptive, redemption, purchase or conversion rights attached to our Common Shares.
As at March 15, 2019, we had 91.64 million Common Shares issued and outstanding.
In addition, as of March 15, 2019 there were 8.35 million Common Shares issuable upon the exercise of outstanding stock options and 3.27 million Common Shares reserved for future grant or issuance under our stock option plan.
We also have 3.52 million Warrants (exercisable into Common Shares) outstanding as at March 15, 2019.
For additional information on stock options and Warrants, please see notes 11 and 12 to our annual consolidated financial statements for the year ended December 31, 2018 which can be retrieved under the Company’s profile on either of the SEDAR or EDGAR websites.
TRADING PRICE AND VOLUME OF AURINIA SHARES
Our Common Shares are listed and posted for trading on the NASDAQ under the symbol “AUPH”, and on the TSX under the symbol “AUP”.
The following table sets forth, for the 12-month period ended December 31, 2018, the reported high and low prices (in United States dollars) and the volume of shares traded for each month on NASDAQ.
NASDAQ |
| | | | | | | | |
| Price Range (US$) | | |
Month | High | | Low | | Total Volume |
January 2018 | 6.11 |
| | 4.52 |
| | 22,658,500 |
|
February 2018 | 5.77 |
| | 4.76 |
| | 12,473,400 |
|
March 2018 | 5.99 |
| | 5.05 |
| | 14,842,415 |
|
April 2018 | 5.65 |
| | 5.01 |
| | 9,900,016 |
|
May 2018 | 6.69 |
| | 5.11 |
| | 15,540,613 |
|
June 2018 | 6.35 |
| | 5.33 |
| | 9,998,308 |
|
July, 2018 | 6.09 |
| | 5.25 |
| | 8,032,718 |
|
August 2018 | 5.90 |
| | 5.22 |
| | 8,509,820 |
|
September 2018 | 6.68 |
| | 5.33 |
| | 11,880,623 |
|
October 2018 | 6.65 |
| | 5.06 |
| | 12,674,871 |
|
November 2018 | 6.15 |
| | 5.10 |
| | 9,753,900 |
|
December 2018 | 7.24 |
| | 5.50 |
| | 16,517,000 |
|
The following table sets forth, for the 12-month period ended December 31, 2018, the reported high and low prices (in Canadian dollars) and the volume of shares traded for each month on the TSX.
TSX
|
| | | | | | | | |
| Price Range (CDN$) | | |
Month | High | | Low | | Total Volume |
January 2018 | 7.58 |
| | 5.70 |
| | 1,344,258 |
|
February 2018 | 7.32 |
| | 5.98 |
| | 854,513 |
|
March 2018 | 7.82 |
| | 6.49 |
| | 973,583 |
|
April 2018 | 7.14 |
| | 6.44 |
| | 535,379 |
|
May 2018 | 8.55 |
| | 6.58 |
| | 1,012,726 |
|
June 2018 | 8.35 |
| | 7.02 |
| | 652,433 |
|
July, 2018 | 8.01 |
| | 6.83 |
| | 464,882 |
|
August 2018 | 7.64 |
| | 6.81 |
| | 433,588 |
|
September 2018 | 8.65 |
| | 6.99 |
| | 677,331 |
|
October 2018 | 8.60 |
| | 6.61 |
| | 908,262 |
|
November 2018 | 8.15 |
| | 6.69 |
| | 839,112 |
|
December 2018 | 9.87 |
| | 7.41 |
| | 1,328,591 |
|
ESCROWED SECURITIES
There are no securities of the Company subject to escrow.
PRIOR SALES
The following table summarizes the distribution of securities other than Common Shares that were issued during the most recently completed financial year, identifying the type of security, the price per security, the number of securities issued, expiry date and the date on which the securities were issued.
STOCK OPTIONS
|
| | | | | | | | | |
Date | Type of Security | | Price per Security (CDN$) | | Number of Securities | | Expiry Date |
February 1, 2018 | Stock Options | | 6.52 |
| | 2,178,000 |
| | February 1, 2028 |
February 5, 2018 | Stock Options | | 6.42 |
| | 550,000 |
| | February 5, 2028 |
February 9, 2018 | Stock Options | | 6.40 |
| | 50,000 |
| | February 9, 2028 |
February 22, 2018 | Stock Options | | 6.92 |
| | 50,000 |
| | February 22, 2028 |
March 21, 2018 | Stock Options | | 7.06 |
| | 150,000 |
| | March 21, 2028 |
October 17, 2018 | Stock Options | | 7.70 |
| | 25,000 |
| | October 17, 2028 |
Total: | | | | | 3,003,000 |
| | |
DIRECTORS AND EXECUTIVE OFFICERS
Our directors are elected by the shareholders at each annual meeting and hold office until the next annual meeting, at which time they may be re-elected or replaced, unless they resign earlier. The executive officers are appointed by the Board and hold office pursuant to individual contractual obligations.
As at March 15, 2019, the names and municipalities of residence of our directors and executive officers and their principal occupations within the five preceding years are set forth below:
|
| | | | | |
Name, province or state, and country of residence | Position with the Company | | Director/Officer since | | Principal Occupation for Five Preceding Years |
Richard Glickman Victoria, British Columbia Canada | Director, Chairman of the Board and CEO | | August 2013 | | CEO of Aurinia since February 2017; Chairman of the Board at Aurinia since August 2013. |
Dennis Bourgeault Edmonton, Alberta Canada | CFO | | May 1998 | | CFO of Aurinia since May 1998. |
Michael R. Martin Victoria, British Columbia Canada | COO | | September, 2013 | | COO of Aurinia since September 2013. |
Neil Solomons Victoria, British Columbia Canada | CMO | | September 2013 | | CMO of Aurinia since September 2013. |
Robert Huizinga North Saanich, British Columbia Canada | Executive Vice President, Corporate Development | | August 2011 | | Executive Vice President, Corporate Development of Aurinia since May 2017; Vice President, Clinical Affairs of Aurinia from August 2011 to May 2017. |
Erik Eglite Lake Forest, Illinois United States
| Senior Vice President, General Counsel & Chief Corporate Compliance Officer | | July 2017 | | Senior Vice President, General Counsel & Chief Corporate Compliance Officer of Aurinia since July 2017; Vice President, Chief Compliance Officer and Corporate Counsel for Marathon Pharmaceuticals and Vice President, Chief Compliance Officer and Corporate Counsel for Lundbeck Pharmaceuticals. Prior to that, Vice President, Chief Compliance Officer and Corporate Counsel for Ovation Pharmaceuticals and Global Chief Compliance Officer, Corporate Counsel for Aspreva Pharmaceuticals. |
Benjamin Rovinski Toronto, Ontario Canada | Director, Chair of the Compensation Committee | | September 2013 | | Managing Director at Lumira Capital, a North American health care and life science venture capital firm. |
David R.W. Jayne Cambridge United Kingdom | Director | | May 2015 | | Certified nephrologist, Director of the Vasculitis and Lupus Clinic and Reader at The University of Cambridge, UK. |
Hyuek Joon Lee Seoul South Korea | Director | | May 2015 | | Managing Director of Business Development for ILJIN Group since December 2016; prior to that, Director of New Business Development for ILJIN Group, a Korean industrial conglomerate; |
Lorin J. ("Jeff") Randall Kennett Square, Pennsylvania United States | Lead Director, Chair of the Audit Committee | | November 2016 | | Corporate director. |
George M. Milne, Jr. Boca Grande, Florida United States | Director, Chair of the Governance and Nominating Committee | | May 2017 | | Corporate director. |
Joseph P, ("Jay") Hagan La Jolla, California United States | Director | | February 2018 | | President and CEO of Regulus Therapeutics Inc., a clinical stage biopharmaceutical company, since May 2017; CFO of Regulus from January 2016 to May 2017; prior thereto held various positions at Orexigen Therapeutics, Inc. and Amgen. |
Michael Hayden Vancouver, British Columbia Canada | Director, Chair of the Standing Research Committee | | February 2018 | | Corporate director; previously President of Global R&D and CSO at Teva Pharmaceutical Industries Limited. |
Directors and executive officers of the Company, as of March 15, 2019, beneficially own, directly or indirectly, 1,913,202 Common Shares in the aggregate, representing 2.09% of the outstanding Common Shares of the Company.
EXECUTIVE OFFICERS AND DIRECTORS
The following are brief biographies of our executive officers and directors.
Richard M. Glickman, LLD (Hon), CEO and Chairman of the Board
Dr. Glickman presently serves as the Company’s CEO and Chairman of the Board. In addition to being a founder of the company, he previously served as the Interim Executive Chairman of the Company for the period September 20, 2013 to February 28, 2014 and as Acting Interim CEO for the period October 22, 2013 to November 5, 2013. He was a co-founder, Chairman and CEO of Aspreva, playing an integral role in the development and establishment of CellCept®, or MMF, as the current standard of care for LN. Aspreva was acquired by Swiss pharmaceutical company Galenica for nearly US$1.0 billion in 2008. He currently serves as founding Chairman of Essa Pharmaceuticals Inc., Chairman of the Board of Engene Corporation and a Director of Correvio Pharma. He is also a Partner at Lumira Capital, one of Canada’s most successful healthcare focused venture capital firms. Dr. Glickman has served on numerous biotechnology and community boards, including member of the federal government’s National Biotechnology Advisory Committee, Director of the Canadian Genetic Disease Network, Chairman of Life Sciences B.C. and a member of the British Columbia Innovation Council. Dr. Glickman is the recipient of numerous awards including the Ernst and Young Entrepreneur of the Year, a recipient of both BC and Canada’s Top 40 under 40 award, the BC Lifesciences 2018 Leadership Award and the 2017 Corporate Leadership Award from the Lupus Foundation of America.
Dennis Bourgeault, CPA-CA, CFO
Dennis Bourgeault has been the CFO of the Company since 1998 and is responsible for the financial and administrative operations of the Company. During his tenure, he contributed significantly to one of the largest Canadian biotechnology PIPE transactions, totaling US$52 million US dollars and was involved in the multi-million-dollar Roche licensing agreement of voclosporin in 2002. In addition, he played a crucial role in executing the merger of Isotechnika and then privately held Aurinia Pharmaceuticals in September 2013. For six years prior to joining Isotechnika, he was the controller for a private industrial distribution company and a Senior Manager in public accounting at KPMG. Mr. Bourgeault obtained his Chartered Accountant designation in 1984.
Michael R. Martin, COO
Michael Martin is currently COO of Aurinia Pharmaceuticals Inc. In this role he oversees all Business Development, Licensing and Partner Management activities along with overall management of the Company's intellectual property portfolio. Additionally, Michael is responsible for the executive leadership of Aurinia's ocular program. Michael was formerly CEO, director and co-founder of the privately held Aurinia Pharmaceuticals Inc., which merged in 2013 with the former Isotechnika Pharma Inc. Michael is a biotech/pharmaceutical executive with over 20 years of industry experience. Michael joined Aurinia from Vifor Pharma where he held the position of Director, Global Business Development & Licensing. Prior to Vifor, Michael was a key member of the business development team that saw Aspreva sold to Galenica for US$915M. Upon joining Aspreva in 2004, Michael initiated the strategic launch planning process for CellCept® in “less-common” autoimmune diseases. These included such indications as pemphigus vulgaris, myasthenia gravis, and lupus nephritis. Prior to this, Michael held a variety of progressively senior commercial positions at Schering-Plough (now Merck). Most recently, he was responsible for the Rheumatology business unit for Remicade® in France. In this role, he had full profit and loss responsibilities and had direct responsibility for the sales team, the marketing team and the infusion access team. In addition, while at Schering-Plough, Michael was the brand manager responsible for the Canadian launch of Remicade (infliximab), which ultimately became the most successful product launch in Canadian history and the largest selling biologic ever. Michael started his career in the industry in the sales organization of Schering-Plough where he received multiple awards and recognition while rapidly progressing towards the prior mentioned roles.
Neil Solomons, MD, CMO
Dr. Neil Solomons co-founded privately-held Aurinia Pharmaceuticals in 2012. He is an experienced pharmaceutical physician with over 20 years of clinical development and medical affairs experience in both large pharma and biotech. He is a recognized expert in rare-disease drug development and is widely published in this field. Neil joined Aurinia from Vifor Pharma, formerly Aspreva Pharmaceuticals (NASDAQ:ASPV) where he held the position of Vice President, Research and Development, being the lead clinician in the development of CellCept® in rare diseases. Neil led the CellCept® Clinical Development teams of over 50 people that saw the completion, reporting, and publication of studies in pemphigus vulgaris and myasthenia gravis (both industry firsts), and the successful landmark lupus nephritis study called ALMS. He was responsible for all clinical development activities from Phases 1 to 3, as well as participating in the formulation of R&D strategy, portfolio management, and due diligence efforts. Prior to Vifor & Aspreva, Neil held a variety of positions at Roche in both Global Clinical Development and Medical Affairs in transplantation, virology, and auto-immune diseases. While at Roche, Dr. Solomons led a diverse team in the development and implementation of post-marketing studies for its transplantation (CellCept® and Zenapax®) and virology (Cytovene®) franchises. Neil qualified in medicine in 1991 receiving his MB BS (MD) at Guys Hospital Medical School, London. He subsequently worked as a physician in London UK, completing specialist training in anesthesia and intensive care. His research interests included sepsis and chronic pain.
Robert B. Huizinga, PhD RN, CNeph(C), Executive Vice President, Corporate Development
Robert Huizinga has more than 25 years of clinical research experience. He has managed the global clinical development of voclosporin since 2002 when he was with Isotechnika Pharma Inc. prior to its merger with Aurinia in 2013. Before joining Isotechnika, Rob was an Investigator in nephrology and transplantation clinical trials where he was involved in more than 60 clinical trials from Phase 1 through Phase 4 and the successful development of numerous compounds including CellCept®, Neoral®, Prograf®, Aranesp® and Simulect®. He has acted as a consultant to nephrology and transplantation pharmaceutical companies, has lectured extensively and is recognized as an expert in immunosuppression drug development. Rob has numerous articles published in leading medical journals, including the Lancet, Kidney International and the American
Journal of Transplantation. He is a member of many professional societies related to nephrology, transplantation, and nursing, has served on many nephrology and transplantation committees and is the founder of RenalPro, a moderated forum for renal professionals. Rob has a PhD (Organizational Leadership) from Regent University, is a Registered Nurse in British Columbia, holds his certification in Nephrology, a M.Sc. in Medicine (Epidemiology) from the University of Alberta, and a member of Sigma Theta Tau (Honor Society of Nursing).
Erik Eglite, DPM, JD, MBA, Senior Vice President, General Counsel & Chief Corporate Compliance Officer
Prior to joining Aurinia, Erik was Vice President, Chief Compliance Officer and Corporate Counsel for Marathon Pharmaceuticals and Vice President, Chief Compliance Officer and Corporate Counsel for Lundbeck Pharmaceuticals. Prior to that, he was Vice President, Chief Compliance Officer and Corporate Counsel for Ovation Pharmaceuticals and Global Chief Compliance Officer, Corporate Counsel for Aspreva Pharmaceuticals. Erik has been involved with the clinical development, launch and commercialization of 15 drugs and drug programs. He is a nationally recognized and frequent speaker on pharmaceutical law. Before entering the pharmaceutical industry, Erik worked as Assistant General Counsel for the Department of Human Services and as a medical malpractice, product liability defense litigation and intellectual property, patent attorney for Querry & Harrow in Chicago, Illinois. He is a licensed podiatric physician and surgeon and is registered to practice before the USPTO, the United States Court of Appeals for the Federal Circuit, the United States Court of Appeals for the District of Columbia Circuit and the United States Seventh Circuit Court of Appeals. Erik has a M.B.A. from the University of Notre Dame. He also holds a B.S. in Biology, a B.A. in History, M.Sc. Cand. in Chemistry, and a J.D. from Loyola University of Chicago. He graduated from Des Moines University Iowa Medical School with a Doctorate in Podiatric Medicine and Surgery, after which he completed his residency training at Michigan Health Medical Center Hospital. He also completed his medical/surgical externships at the University of Chicago, Department of Surgery, Division of Vascular Surgery and Northwestern University Columbus Cabrini Hospital, Department of Orthopedic/Podiatric Surgery. He has a graduate certificate in Pharmaceutical & Medical Device Law from Seton Hall School of Law, an Executive Certificate in Corporate Governance from Northwestern University Kellogg School of Management and an Executive Certificate in Business Administration from the University of Notre Dame. Currently, he is completing his M.S. in Regulatory Compliance at Northwestern University.
Lorin Jeffry Randall, MBA, Lead Director, Chair of the Audit Committee
Mr. Jeff Randall has over 30 years of experience serving in financial and operating roles spanning biotechnology, pharmaceuticals and manufacturing. He has led a number of companies through multi-million-dollar financings and mergers and acquisitions. In addition to his current board positions, Mr. Randall served on the board of directors of Nanosphere, Inc. from 2008 to 2016, most recently as Chairman of the Board. From 2004 to 2006, Mr. Randall, a financial consultant, was Senior Vice President and CFO of Eximias Pharmaceutical Corporation, a development-stage drug development company. Mr. Randall holds a Bachelor of Science in Mathematics and Accounting from Pennsylvania State University and a Master’s in Business Administration from Northeastern University.
Benjamin Rovinski, PhD, Director, Chair of the Compensation Committee
Dr. Benjamin Rovinski has 30 years of investment, operational, managerial and research experience in the healthcare sector. Dr. Rovinski joined Lumira Capital in 2001, where he is a Managing Director, with an investment focus on early- to late-stage private and public life sciences companies. Prior to joining Lumira Capital, he held several senior management positions in the biotechnology sector, including 13 years at Sanofi Pasteur where he was a senior scientist and director of molecular virology. Dr. Rovinski led global R&D programs in the areas of HIV/AIDS and therapeutic cancer vaccines, bringing several of them through to clinical-stage. Dr. Rovinski holds a Ph.D. in Biochemistry from McGill University in Montréal and did post-doctoral studies in Molecular Oncology and Retrovirology at the Ontario Cancer Institute in Toronto. He obtained his undergraduate degree from Rice University in Houston. His current and past board roles and investment responsibilities include several private and public companies, including Antios Therapeutics; Antiva Biosciences; GI Therapeutics (NASDAQ:GTHX); Vascular Pharmaceuticals; KAI Pharmaceuticals (acquired by Amgen); Morphotek (acquired by Eisai); Cervelo Pharmaceuticals; Health Hero Network (acquired by Bosch); Avalon Pharmaceuticals (NASDAQ: AVRX; acquired by Clinical Data, Inc.); Inovise Medical, Inc.; Protana; Signature Biosciences; and SGX Pharmaceuticals (NASDAQ: SGXP; acquired by Eli Lilly). He also serves on the board of directors of Life Sciences Ontario, Ontario Genomics, and the steering committee of the Toronto Regional Board of Trade’s Health Science Cluster initiative. Dr. Rovinski has published over 25 scientific articles and reviews and is the recipient of 31 issued patents.
David R.W. Jayne, MD, FRCP, FRCPE, FmedSci, Director
Dr. David Jayne is Professor of Clinical Autoimmunity in the Department of Medicine at the University of Cambridge, UK. Dr. Jayne received his MB BChir in Surgery and Medicine from Cambridge University, Cambridge, England. He received postgraduate training at several London hospitals and Harvard University. He is a fellow of the Royal Colleges of Physicians of London and Edinburgh, and the Academy of Medical Science. He is a certified nephrologist and an Honorary Consultant Physician at Addenbrooke’s Hospital, Cambridge UK. Dr. Jayne is a medical advisor to UK, U.S. and EU regulatory bodies, patient groups and professional organizations. He has published more than 400 peer-reviewed journal articles, book chapters and reviews. He was elected the first President of the European Vasculitis Society in 2011 and is a member of the ERA-EDTA immunopathology working group and he co-chairs the EULAR/ERA-EDTA task force on lupus nephritis. Dr. Jayne’s research includes investigator-initiated international trials and the introduction of newer therapies in vasculitis and SLE with collaborators on five continents.
Hyuek Joon Lee, PhD, Director
Dr. Joon Lee is the Managing Director of Business Development for ILJIN Group and is responsible for mergers and acquisitions, and managing overseas investments, joint ventures and subsidiaries. As of October 2014, he joined the board of directors of Life Science Enterprises in Massachusetts, a privately held company focusing on advanced biomaterials that promote bone repair. Dr. Lee has over 20 years of experience in consulting, management, business development and strategic planning in a number of industries including information technology, chemical
and media. Dr. Lee received his B.S. in Chemistry from Seoul National University, and his M.S.E. and Ph.D. in Chemical Engineering from the University of Michigan, Ann Arbor.
George M. Milne, Jr., PhD, Director, Chair of the Governance & Nomination Committee
Dr. Milne has over 30 years of experience in pharmaceutical research and product development. Dr. Milne currently serves on the boards of Amylyx Pharmaceuticals, Inc. and Charles River Laboratories, Inc. where he is the lead director. He has retired from Pfizer where he served as Executive Vice President of Global Research and Development and President, Worldwide Strategic and Operations Management. He joined Pfizer in 1970 and held a variety of positions conducting both chemistry and pharmacology research. Dr. Milne became director of the department of immunology and infectious diseases at Pfizer in 1981, was its executive director from 1984 to 1985, and was vice president of research and development from 1985 to 1988. He was appointed senior vice president in 1988. In 1993 he was appointed President of Pfizer Central Research and a senior vice president of Pfizer Inc. with global responsibility for human and veterinary medicine research and development. Dr. Milne has served on multiple corporate boards including Mettler-Toledo, Inc. (a manufacturer of laboratory instruments), MedImmune, Athersys, Biostorage Technologies, Aspreva and Conor Medsystems. Dr. Milne received his B.Sc. in Chemistry from Yale University and his Ph.D. in Organic Chemistry from MIT.
Joseph P. Hagan, MBA, Director
Mr. Hagan is President and CEO of Regulus Therapeutics. Mr. Hagan joined Regulus in January 2016 as COO, Principal Financial Officer and Principal Accounting Officer and was appointed to President and CEO in May 2017. Mr. Hagan’s career includes roles as the Executive Vice President, CFO and Chief Business Officer of Orexigen Therapeutics, Inc., Managing Director of Amgen Ventures and head of corporate development for Amgen Inc. Mr. Hagan has led numerous strategic and financing transactions including the acquisitions of Immunex and Tularik and the spinout of Novantrone and Relyspa, as well as many other business development efforts totaling over US$15 billion in value. Before joining Amgen, Mr. Hagan spent five years in the bioengineering labs at Genzyme and Advanced Tissue Sciences. Mr. Hagan currently serves on the board of directors of Zosano Pharma, a publicly traded biotechnology company. He received an M.B.A. from Northeastern University and a B.S. in Physiology and Neuroscience from the University of California, San Diego.
Michael Hayden, CM, OBC, MB, ChB, PhD, FRCP(C), FRSC, Director, Chair of the Standing Research Committee
Dr. Michael Hayden was recently named one of the 50 Canadians born in the 20th century who have changed the world. He is the co-founder of five biotechnology companies: Prilenia Therapeutics, 89Bio, NeuroVir Therapeutics Inc., Xenon Pharmaceuticals Inc., and Aspreva Pharmaceuticals Corp. Dr. Hayden sits on different boards including Xenon Pharmaceuticals and Ionis Pharmaceuticals. Author of over 860 peer-reviewed publications and invited submissions, Dr. Hayden has focused his research primarily on genetic diseases, including genetics of diabetes, lipoprotein disorders, Huntington disease, predictive and personalized medicine. Dr. Hayden was inducted into the Canadian Medical Hall of Fame in 2017. He was named one of PharmaVoice’s “100 of the Most Inspiring People” (2015); awarded an Honorary Doctor of Science by the University of Gottingen (2014); the Luminary award by the Personalized Medicine World Conference (2014); and the Diamond Jubilee Medal (2012), on behalf of HRH Queen Elisabeth II, in recognition of his significant contributions and achievements. Dr. Hayden has also been awarded the Order of Canada (2011), and the Order of British Columbia (2010). He was named Canada’s Health Researcher of the Year by CIHR (NIH of Canada) in 2008, and he received the Prix Galien in 2007, which recognizes the outstanding contribution of a researcher to Canadian pharmaceutical research.
COMMITTEES OF THE BOARD
We have four standing committees: the Audit Committee, the Governance and Nomination Committee, the Compensation Committee, and Standing Research Committee. Current members of these committees are identified in the following table:
|
| |
Committee | Members |
Audit Committee (1) | Lorin J. Randall (Chair) Benjamin Rovinski Hyuek Joon Lee |
Governance and Nomination Committee | George M. Milne, Jr. (Chair) David Jayne Hyuek Joon Lee |
Compensation Committee | Benjamin Rovinski (Chair) Lorin J. Randall Hyuek Joon Lee |
Standing Research Committee | Michael Hayden (Chair) David Jayne |
| |
(1) | Detailed information on the Audit Committee is attached as Schedule 1. |
CEASE TRADE ORDERS, BANKRUPTCIES, PENALTIES OR SANCTIONS
No director or executive officer of the Company is, or has been within 10 years before the date of this AIF, a director, chief executive officer or chief financial officer of any company, including Aurinia, that:
| |
(a) | was subject to a cease trade order, an order similar to a cease trade order or an order that denied the relevant company access to any exemption under securities legislation, that was issued while the proposed director was acting in the capacity as a director, chief executive officer or chief financial officer; or |
| |
(b) | was subject to a cease trade order, an order similar to a cease trade order or an order that denied the relevant company access to any exemption under securities legislation, that was issued after the director or executive officer ceased to be a director, chief executive officer or chief financial officer and which resulted from an event that occurred while he was acting in the capacity of a director, chief executive officer or chief financial officer. |
No director or executive officer of the Company, except as noted below, or shareholder holding a sufficient number of securities of the Company to affect materially the control of the Company:
| |
(a) | is, or has been within 10 years before the date of this AIF, a director, chief executive officer or chief financial officer of any company, including Aurinia, that while that person was acting in that capacity, or within a year of that person ceasing to act in that capacity, became bankrupt, made a proposal under any legislation relating to bankruptcy or insolvency, or was subject to or instituted any proceedings, arrangement or compromise with creditors, or had a receiver, receiver manager or trustee appointed to hold its assets; or |
| |
(b) | has, within 10 years before the date of this AIF, become bankrupt, made a proposal under any legislation relating to bankruptcy or insolvency, or become subject to or instituted any proceedings, arrangement or compromise with creditors, or had a receiver, receiver manager or trustee appointed to hold the assets of the director, chief executive officer or chief financial officer. |
Mr. Lorin J. Randall was a director of Tengion, Inc. when it filed for Chapter 7 Liquidation in December of 2015 pursuant to the United States Bankrupcy Code.
No director or executive officer of the Company, or shareholder holding a sufficient number of securities of the Company to affect materially the control of the Company, has been subject to:
| |
(a) | any penalties or sanctions imposed by a court relating to securities legislation or by a securities regulatory authority or has entered into a settlement agreement with a securities regulatory authority; |
| |
(b) | any other penalties or sanctions imposed by a court or regulatory body that would likely be considered important to a reasonable investor in making an investment decision. |
LEGAL PROCEEDINGS AND REGULATORY ACTIONS
As of March 15, 2019, we are not aware of any legal proceedings against us that would involve a claim for damages that exceed ten per cent of our current assets.
No penalties or sanctions have been imposed against us by a court relating to securities legislation or any securities regulatory authority during the financial year ended December 31, 2018, nor have we entered into any settlement agreements with a court relating to securities legislation or with a securities regulatory authority during such financial year. No other penalties or sanctions have been imposed by a court or regulatory body against us which would likely be considered important to a reasonable investor in making an investment decision respecting the Company.
INTEREST OF MANAGEMENT AND OTHERS IN MATERIAL TRANSACTIONS
None of our directors or executive officers, persons or companies that beneficially own, control, or direct more than 10% of our voting securities, or an associate or affiliate of any of such directors, executive officers, persons or companies, had a material interest, directly or indirectly, in the transactions conducted by the Company within the three most recently completed financial years or during the current financial year that has materially affected or is reasonably expected to materially affect the Company.
CONFLICTS OF INTEREST
To our knowledge, and other than as disclosed herein, there is no known existing or potential material conflicts of interest among the Company, its directors and officers, or a subsidiary of the Company or other members of management as a result of their outside business interests, except
that certain of its directors may serve as directors of other companies and therefore it is possible that a conflict may arise between their duties to the Company and their duties as a director of such other companies. See “Risk Factors - The Company is dependent upon its key personnel to achieve its business objectives”.
TRANSFER AGENT AND REGISTRAR
Our co-transfer agents and co-registrars are Computershare Investor Services Inc. located at its principal offices in Calgary, Alberta and Toronto, Ontario and Computershare Trust Company, N.A. located at its principal offices in Golden, Colorado.
MATERIAL CONTRACTS
We currently have the following material contracts:
| |
1. | Under the terms of an agreement dated February 14, 2014 between the Company and Dr. Robert Foster, whereby Dr. Robert Foster’s employment as CSO was terminated by the Company, it was confirmed that effective March 8, 2012, Dr. Foster was entitled to receive 2% of royalty licensing revenue for royalties received on the sale of voclosporin by licensees and/or 0.3% of net sales of voclosporin sold directly by the Company, to be paid quarterly as that revenue is received by the Company. Should the Company sell substantially all of the assets of voclosporin to a third party or transfer those assets to another party in a merger in a manner such that this payment obligation is no longer operative, then Dr. Foster will be entitled to receive 0.3% of the value attributable to voclosporin in the transaction. |
| |
2. | The manufacturing collaboration and services agreement, dated November 22, 2016 between Lonza and the Company as described under the heading “Manufacturing, Encapsulating and Packaging of Voclosporin - Lonza Manufacturing Collaboration Agreement”. |
INTERESTS OF EXPERTS
PricewaterhouseCoopers LLP, the Company’s auditor, issued an auditor’s report dated March 15, 2019 in respect of our Consolidated Financial Statements, which comprise the Consolidated Statements of Financial Position as at December 31, 2018 and December 31, 2017, and the Consolidated Statements of Operations and Comprehensive Loss, Consolidated Statements of Changes in Shareholders’ Equity and Cash Flows for the years ended December 31, 2018 and December 31, 2017, and the related notes. PricewaterhouseCoopers LLP has advised us that they are independent with respect to the Company within the meaning of the Rules of Professional Conduct of the Chartered Professional Accountants of Alberta and the rules of the SEC.
ADDITIONAL INFORMATION
Additional information with respect to the Company, including directors’ and officers’ remuneration and indebtedness, principal holders of our Common Shares and securities authorized for issuance under equity compensation plans will be contained in the most recently filed management information circular of the Company. Additional financial information is also available in our comparative audited consolidated financial statements, together with the auditor’s report thereon, and the related Management Discussion and Analysis for its most recently completed fiscal year ended December 31, 2018.
Additional information regarding the Company is available on the SEDAR website located at www.sedar.com, on EDGAR at www.sec.gov/edgar, or on the Company’s corporate website located at www.auriniapharma.com, or upon request addressed to Michael Martin, COO, at 1203, 4464 Markham Street, Victoria, British Columbia V8Z 7X8.
SCHEDULE 1 - AUDIT COMMITTEE INFORMATION
| |
1. | The Audit Committee’s Charter |
Our Audit Committee Charter is available in the governance section of our website at www.auriniapharma.com and is attached as Schedule 2 to this AIF.
| |
2. | Composition and Relevant Education and Experience |
The Audit Committee is comprised of three independent directors: Lorin J. Randall (Chair), Benjamin Rovinski and Hyuek Joon Lee. A description of the education and experience of each Audit Committee member that is relevant to the performance of his responsibilities as an Audit Committee member may be found above under the heading “Directors and Executive Officers”.
Under the SEC rules implementing the Sarbanes-Oxley Act of 2002, Canadian issuers filing reports in the United States must disclose whether their audit committees have at least one audit committee financial expert. The Board has determined that Lorin J. Randall qualifies as an audit committee financial expert under such rules. In addition, all members of the Audit Committee are considered financially literate under applicable Canadian and U.S. laws.
| |
3. | Pre-approval Policies and Procedures |
The Audit Committee is authorized by the Board to review the performance of our external auditor and approve in advance the provision of services other than auditing and to consider the independence of the external auditor, including reviewing the range of services provided in the context of all consulting services bought by us. Such advance approval authority may be delegated by the Audit Committee to the Chair of the Audit Committee who is “independent” and “unrelated”.
All fees for audit and audit related services performed by the external auditor for the year ended December 31, 2018 were pre-approved by the Audit Committee. All fees for non-audit related services performed by the external auditor for the year ended December 31, 2018 were pre-approved by the Audit Committee and/or Audit Chair as delegated by the Audit Committee.
| |
4. | External Auditor Service Fees (By Category) |
The aggregate fees recorded for professional services rendered by the external auditor, PricewaterhouseCoopers LLP, for the Company and its subsidiaries for the years ended December 31, 2018 and 2017, respectively are as follows:
|
| | | | | | | | | | | | | |
Fiscal year ended | 2018 | | % of Total Fees | | 2017 | | % of Total Fees |
Audit fees (for audit of the Company’s annual financial statements and services provided in connection with statutory and regulatory filings)(1) | $ | 95,124 |
| | 31.5 | % | | $ | 130,583 |
| | 50.2 | % |
Audit related fees, including review of the Company’s quarterly financial statements(2) | $ | 40,825 |
| | 13.5 | % | | $ | 37,491 |
| | 14.4 | % |
Tax fees (tax compliance, tax advice and planning)(3) | $ | 110,496 |
| | 36.6 | % | | $ | 44,935 |
| | 17.3 | % |
All other fees(4) | $ | 55,647 |
| | 18.4 | % | | $ | 46,943 |
| | 18.1 | % |
Total fees | $ | 302,092 |
| | 100.0 | % | | $ | 259,952 |
| | 100.0 | % |
| |
(1) | These fees include professional services provided by the external auditor for the statutory audits of the annual financial statements. The total for 2018 is comprised of US$56,910 related to interim billings for the 2018 audit and US$38,214 related to fees for the 2017 audit billed in 2018. The total for 2017 is comprised of US$61,688 related to interim billings for the 2017 audit and US$68,895 related to fees for the 2016 audit billed in 2017. |
| |
(2) | These fees relate to performing review engagement services on the Company’s quarterly financial statements and other audit related services. |
| |
(3) | These fees include professional services for transfer pricing, tax compliance, tax advice, tax planning and various taxation matters. |
| |
(4) | These fees for 2018 include professional services for assistance in filing the new base shelf prospectus, prospectus supplement related to the re-sale of common shares, the November 2018 ATM prospectus supplement, and various other advisory services. These fees for 2017 include professional services for assistance in filing prospectus supplements for the December 2016 bought deal financing and the March 20, 2017 public offering, and the new preliminary base shelf prospectus. |
SCHEDULE 2 - AUDIT COMMITTEE CHARTER
AURINIA PHARMACEUTICALS INC.
AUDIT COMMITTEE CHARTER
PURPOSE
The purpose of the Audit Committee of the Board of Directors of Aurinia Pharmaceuticals Inc. (the “Company”) shall be to assist the Board of Directors of the Company (the “Board”) in its oversight of (i) the quality and integrity of the financial statements of the Company, (ii) the Company’s compliance with legal and regulatory requirements, (iii) the accounting and financial management processes of the Company, and the effectiveness of the Company’s internal controls over financial reporting, (iv) the quality and integrity of the annual audit of the Company’s financial statements, including the independence and qualifications of the Company’s independent auditor.
MEMBERSHIP
The Committee shall consist of no fewer than three (3) members. None of the members of the Committee shall be an officer or employee of the Company or any of its subsidiaries, and each member of the Committee shall be an independent director (in accordance with the definition of “independent director” and “independent” established from time to time under the requirements or guidelines for audit committee service under applicable securities laws (“Securities Laws”) and the rules of any stock exchange (“Exchange Rules”) on which the Company’s shares are listed for trading).
| |
2. | Appointment and Replacement of Committee Members |
Any member of the Committee may be removed or replaced at any time by the Board and shall automatically cease to be a member of the Committee upon ceasing to be a director. The Board may fill vacancies on the Committee by election from among its members. The Board shall fill any vacancy if the membership of the Committee is less than three directors. If and whenever a vacancy shall exist on the Committee, the remaining members may exercise all its power so long as a quorum remains in office. Subject to the foregoing, the members of the Committee shall be elected by the Board annually and each member of the Committee shall hold office as such until the next annual meeting of shareholders after his or her election or until his or her successor shall be duly elected and qualified.
All members of the Committee should be “financially literate” (as that term may be defined from time to time under the requirements or guidelines for audit committee service under applicable Securities Laws and the Exchange Rules) or must become financially literate within a reasonable period of time after his or her appointment to the Committee.
In addition, at least one member must have past employment experience in finance or accounting, requisite professional certification in accounting or any other comparable experience or background which results in the individual’s financial sophistication. Unless otherwise determined by the Board, at least one member of the Audit Committee shall be an “audit committee financial expert”.
RESPONSIBILITIES AND DUTIES
The principal responsibilities and duties of the Committee in serving the purposes outlined above in this charter are set forth below. These duties are set forth as a guide with the understanding that the Committee will carry them out in a manner that is appropriate given the Company’s needs and circumstances. The Committee may supplement them as appropriate and may establish policies and procedures from time to time that it deems necessary or advisable in fulfilling its responsibilities.
1.Appointment and Oversight of Independent Auditor. The Committee recommends to the Board for nomination the independent auditor to examine the Company’s accounts, controls and financial statements. The Committee has sole responsibility for the compensation, retention, oversight and, if necessary, termination of any independent auditor (including resolution of disagreements between the Company’s management and the independent auditor regarding financial reporting) for the purpose of preparing or issuing an audit report or performing other audit, review or attest services for the Company, and the independent auditor and will report directly to the Committee.
| |
2. | Auditor Independence and Qualifications |
(a)The Committee is responsible for assessing the independent auditor’s qualifications, performance and independence annually, and for taking, or recommending that the full Board take, appropriate action to oversee the independence of the independent auditor.
In connection therewith, the Committee will:
(i)make sure it reviews, on an annual basis, all relationships between the independent auditor and the Company, including those described in the formal written statement that the Committee obtains annually from the independent auditor under applicable requirements of the Canadian generally accepted auditing standards (CAS) and the Public Company Accounting Oversight Board (the “PCAOB”) related to the independent auditor’s communications with the Committee concerning independence; and
(ii)actively engage in a dialogue with the independent auditor with respect to any disclosed relationships or services that may impact the objectivity and independence of the independent auditor.
(b)The Committee will obtain and review, at least annually, a report from the independent auditor describing:
(i)the independent auditor’s internal quality-control procedures; and
(ii)any material issues raised by the most recent internal quality-control review, peer review Canadian Public Accountability Board (CPAB) or PCAOB review of the independent auditor, or by any governmental or professional authority in any inquiry or investigation, within the preceding five years, regarding any independent audit carried out by the independent auditor, and any steps taken to address any such issues.
(c)The Committee is responsible for reviewing and evaluating the lead audit partner of the independent auditor and overseeing the rotation of the lead audit partner as required by applicable law. In
making its evaluation, the Committee should take into account the opinions of management and the independent auditor.
(d)The Committee will set policies for the Company’s hiring of employees or former employees of the present and former independent auditor.
3.Approval of Audit and Non-Audit Services
(a)The Committee will review the independent auditor’s audit planning, scope and staffing.
(b)The Committee must pre-approve all audit and non-audit related services provided to the Company by the independent auditor. The Committee may establish pre-approval policies and procedures, as permitted by the Exchange Rules, Securities Laws and applicable law, for the engagement of the independent auditor to render services to the Company, including, without limitation, policies that would allow the delegation of pre-approval authority to one or more members of the Committee, provided that any pre-approval decision is reported to the Committee at its next scheduled meeting.
4.Interaction with Independent Auditor
(a)The Committee will, to the extent warranted, discuss with the independent auditor the reports referenced in section 2(b) and any other matters required to be reviewed under applicable legal and regulatory requirements.
(b)The Committee will periodically consult with the independent auditor, out of the presence of the Company’s management, about the Company’s internal controls, the fullness and accuracy of the Company’s financial statements, the responsibilities, budget and staffing of the Company’s finance function, and any other matters that the Committee or independent auditor believes should be discussed privately out of the presence of management.
| |
B. | FINANCIAL STATEMENTS AND DISCLOSURES |
1.Annual Financial Statements and Disclosures
(a)Before public disclosure, the Committee will meet to review and discuss with the independent auditor and the Company’s management the Company’s audited consolidated financial statements and the notes and Managements’ Discussion and Analysis relating to such consolidated financial statements, the annual report, the annual information form, the financial information of the Company contained in any prospectus or information circular or other disclosure documents or regulatory filings of the Company, the recommendations for approval of each of the foregoing from each of the President and Chief Executive Officer, and Chief Financial Officer of the Company and based on such recommendations provide, where applicable, its own recommendations to the Board for their approval and release of each of the foregoing to the public.
(b)The Committee will discuss with the independent auditor and the Company’s management any items appropriate or required to be discussed in accordance with applicable auditing and CPAB standards in connection with the preparation of the Company’s annual financial statements, including any problems or difficulties encountered during the course of the audit, including any restrictions on the scope of work or access to required information, and any significant disagreements with management and management’s response to such difficulties.
2.Quarterly Financial Statements and Disclosures
(a)The Committee will meet to review and discuss with the independent auditor and the Company’s management the Company’s interim consolidated financial statements and the notes and Managements’ Discussion and Analysis relating to such consolidated financial statements before public disclosure, and either, in the discretion of the Audit Committee, (A) approve and release each of the foregoing to the public, or (B) provide, where applicable, its own recommendation to the Board for their approval and release of each of the foregoing to the public.
(b)The Committee will discuss with the independent auditor and the Company’s management any items appropriate or required to be discussed in accordance with applicable auditing and CPAB standards in connection with the preparation of the Company’s quarterly financial statements.
3.Earnings Announcements and Guidance. The Committee will discuss generally with the Company’s management and the independent auditor, as appropriate, the type of information to be disclosed and type of presentation to be made regarding the Company’s earnings press releases.
4.Ongoing Reviews. In connection with the foregoing, the Committee will review the Company’s financial reporting and accounting standards and principles and financial statement presentations, significant changes in the selection of such standards or principles or in their application and the key accounting decisions affecting the Company’s financial statements, including alternatives to, and the rationale for, the decisions made. As part of this review, the Committee will discuss with the Company’s management and the independent auditor the reasonableness of judgments and estimates used in the preparation of financial statements, and alternative accounting treatments, principles or practices that were considered or may be preferred by the independent auditor, the Committee or the Company’s management.
| |
C. | CONTROLS AND PROCEDURES |
1.Review of Processes, Systems, Controls and Procedures. The Committee will periodically review and meet separately with the independent auditor, or other personnel primarily responsible for the internal control, and the Company’s management to discuss their periodic reviews of the integrity, adequacy and effectiveness of the Company’s accounting and financial reporting processes, systems of internal control (including any significant deficiencies and material weaknesses in their design or operation), and disclosure controls and procedures (and management’s reports thereon), as well as any special audit steps adopted in light of material control deficiencies. The Committee shall receive and review the required applicable annual or quarterly CEO and CFO certification reports prior to these documents being filed as required by the regulators.
(a)The Committee will periodically review with the Company’s management and the Company’s legal counsel, the nature and status of significant legal matters.
(b)The Committee will review and monitor any significant pending or threatened litigation that could have a material impact on the Company’s financial statements.
3.Risk Assessment and Risk Management. The Committee is responsible for overseeing the management of risks associated with the Company’s financial reporting, accounting and auditing matters, reviewing as required the Company’s processes around the management and monitoring of such risks, including but not limited to, review and assessment of the company investment policy and performance
and review and assessment of the company’s insurance policies. The Committee will discuss with the Company’s management the Company’s major financial, accounting and reporting risk exposures and the steps management has taken to monitor and control such exposures, including the Company’s risk assessment and risk management policies and guidelines.
4.Whistleblower Procedures. The Committee is responsible for establishing and overseeing procedures for the receipt, retention and treatment of complaints received by the Company regarding accounting, internal accounting controls or auditing matters, the prompt internal reporting of violations of the Code of Business Conduct and Ethics and for the confidential, anonymous submission by Company employees of concerns regarding questionable accounting or auditing matters.
| |
D. | OTHER DUTIES AND RESPONSIBILITIES |
1.Code of Conduct. The Committee will periodically review and recommend to the Board any changes to the Code of Conduct applicable to the Company, including all of its directors, officers and employees. The Committee will also consider waivers of the Code of Conduct requested for executive officers and directors and retain sole authority to grant any waivers for executive officers and directors (other than where the potential waiver involves a member of the Committee, in which event such waiver shall be subject to the review of the Board). The Committee will also periodically review and recommend to the Board any changes to the Company’s Insider Trading Policy and Anti-Bribery Policy, which are referenced in the Company’s Code of Conduct.
| |
2. | Related Party Transactions. The Committee will review and, where appropriate, approve any transaction between the Company and any related party (other than transactions that are subject to review by the Board as a whole or any other committee of the Board), as defined |
by applicable law, Securities Laws and the Exchange Rules, and will periodically review the business interests and activities of members of the Board and management.
3.Review of Composition and Performance. The Committee will evaluate the Committee’s composition and performance on an annual basis and submit a report to the Board.
4.Review of this Charter. The Committee will review and reassess the adequacy of this charter annually and recommend to the Board any changes the Committee determines are appropriate.
5.Other Actions. The Committee will perform any other activities required by applicable law, rules or regulations, including Securities Laws and the Exchange Rules, and take such other actions and perform and carry out any other responsibilities and duties delegated to it by the Board or as the Committee deems necessary or appropriate consistent with its purpose.
In discharging its responsibilities, the Committee may conduct, direct, supervise or authorize studies of, or investigations into, any matter that the Committee deems appropriate, with full and unrestricted access to all books, records, documents, facilities and personnel of the Company. The Committee has the sole authority to retain and terminate independent legal counsel and other consultants, accountants, experts and advisers of its choice to assist the Committee in connection with its functions, including any studies or investigations. The Committee will have the sole authority to approve the fees and other retention terms of such advisers. The Company will also provide for appropriate funding, as determined by the Committee, for:
| |
• | payment of compensation to the independent auditor and any legal and other consultants, accountants, experts and advisers retained by the Committee; and |
| |
• | ordinary administrative expenses of the Committee that are necessary and appropriate in carrying out its functions. |
Meetings of the Committee shall be held at least once each quarter or more frequently, as determined to be appropriate by the Committee. The Board may appoint a member of the Committee to serve as the chairperson of the Committee (the “Chair”); if the Board does not appoint a Chair, the Committee members may designate a Chair by their majority vote. The Chair, in consultation with the other members of the Committee, will set the dates, time, places and agenda for Committee meetings. The Chair or any other member of the Committee may call meetings of the Committee by notice and the Committee may act by unanimous written consent in lieu of a meeting in accordance with the Company’s Bylaws. A quorum of the Committee for the transaction of business will be a majority of its members. Meetings may be held in person or via telephone or video conference. The Committee also may act by unanimous written consent in lieu of a meeting in accordance with the Company’s Bylaws. Subject to the requirements of this charter, applicable law, Securities Laws and the Commission Rules, the Committee and the Chair may invite any director, executive or employee of the Company, or such other person, as it deems appropriate in order to carry out its responsibilities, to attend and participate (in a non-voting capacity) in all or a portion of any Committee meeting. The Committee may meet in executive session at its discretion and may exclude from all or a portion of its meetings any person it deems appropriate in order to carry out its responsibilities. The Chair will designate a secretary for each meeting, who need not be a member of the Committee. The Company shall provide the Committee such staff support as it may require.
The Committee will maintain written minutes of its meetings and copies of its actions by written consent, and will cause such minutes and copies of written consents to be filed with the minutes of the meetings of the Board. The Committee will report regularly to the Board with respect to its activities, including on significant matters related to the Committee’s responsibilities and the Committee’s deliberations and actions. The minutes of the Committee and actions by the unanimous written consent of the Committee members will be made available to the other members of the Board.
| |
H. | DELEGATION OF AUTHORITY |
The Committee may from time to time, as it deems appropriate and to the extent permitted under applicable law, Securities Laws and the Commission Rules, the Company’s articles of incorporation and Bylaws, form and delegate authority to subcommittees.
Members of the Committee will receive such fees, if any, for their service as Committee members as may be determined by the Board, which may include additional compensation for the Chair. Such fees may include retainers or per meeting fees and will be paid in such form of consideration as is determined by the Board in accordance with applicable law, Securities Laws and the Commission Rules.
The Company shall make this charter freely available to stockholders on request and shall publish it on the Company’s web site.
This charter sets forth the authority and responsibility of the Committee in fulfilling the purposes described herein.
While the Committee has the responsibilities and powers set forth in this Charter, it is not the duty of the Committee to plan or conduct audits or to determine that the Company’s consolidated financial statements are complete and accurate or are in accordance with International Financial Reporting Standards (“IFRS”) and applicable rules and regulations. These are the responsibilities of Management and the Company’s external auditors. The Committee, its Chair and any Committee members identified as having accounting or related financial expertise are members of the Board, appointed to the Committee to provide broad oversight of the financial, risk and control related activities of the Company, and are specifically not accountable or responsible for the day-to-day operation or performance of such activities. Although the designation of a Committee member as having accounting or related financial expertise for disclosure purposes or otherwise is based on that individual’s education and experience which that individual will bring to bear in carrying out his or her duties on the Committee, such designation does not impose on such person any duties, obligations or liability that are greater than the duties, obligations and liability imposed on such person as a member of the Committee and Board in the absence of such designation. Rather, the role of a Committee member who is identified as having accounting or related financial expertise, like the role of all Committee members, is to oversee the process, not to certify or guarantee the internal or external audit of the Company’s financial information or public disclosure.
In addition, the Company’s management is responsible for managing its risk function and for reporting on its processes and assessments with respect to the Company’s management of risk. Each member of the Committee shall be entitled to rely on (a) the integrity of those persons and organizations within and outside of the Company from which it receives information, (b) the accuracy of the financial and other information provided to the Committee by such persons or organizations absent actual knowledge to the contrary (which shall be promptly reported to the Board) and (c) representations made by management as to any audit and non-audit services provided by the independent auditor.
The Board has formed the Committee to assist the Board in directing the Company’s affairs and this charter has been adopted in furtherance of this purpose. While this charter should be interpreted in the context of all applicable laws, regulations and listing requirements, as well as in the context of the Company’s articles of incorporation and Bylaws, it is not intended to establish by its own force any legally binding obligations.
SCHEDULE 3 - GLOSSARY OF TERMS AND DEFINITIONS
In this annual information form, the following capitalized words and terms shall have the following meanings:
"2015 Base Shelf Prospectus" means Aurinia's short form base shelf prospectus dated October 16, 2015;
"AEs" means adverse events;
"AIF" means the Annual Information Form of the Company dated March 15, 2019 for the fiscal year ended December 31, 2018;
"ALMS" means the Aspreva Lupus Management Study;
"Anti-dsDNA" means double-stranded DNA;
"API" means active pharmaceutical ingredient;
"Aspreva" means Aspreva Pharmaceuticals Inc.;
"ATM" means an At-the-Market Facility;
"AURA-LV (AURA)" means a Phase 2b clinical trial. The protocol is titled "A Randomized, Controlled Double-blind Study Comparing the Efficacy and Safety of Voclosporin (23.7 mg BID, or 39.5 mg BID) with Placebo in Achieving remission in Patients with Active Lupus Nephritis";
"Aurinia" means Aurinia Pharmaceuticals Inc.;
"AURION" means an open label exploratory study. The protocol is titled "An Exploratory study assessing the Short term Predictors of Remission of Voclosporin 23.7 mg BID in combination with standard of care in Patients with Active Lupus Nephritis";
"AURORA" means a single double-blind, randomized, placebo controlled Phase 3 clinical trial for voclosporin in the treatment of LN;
"AURORA 2 extension trial" means a 104-week blinded extension trial;
"BID" means administered twice a day;
"Board" means the board of directors of the Company;
"calcineurin" means a specific enzyme (phosphatase enzyme) that can have its activity inhibited by immunosuppressive (anti-organ rejection) drugs, including, for example, cyclosporine;
"Catalent" means Catalent Pharma Solutions;
"CellCept®" means the brand name of MMF;
"CEO" means Chief Executive Officer;
"CFO" means Chief Financial Officer;
"CMO" means Chief Medical Officer;
"CNI" means calcineurin inhibitors, the cornerstone of therapy for the prevention of organ transplant rejection;
"Company" means Aurinia Pharmaceuticals Inc. and (unless the context specifies or implies otherwise) its subsidiaries;
"Common Shares" means common shares in the authorized share capital of the Company;
"COO" means Chief Operating Officer;
"CR" means complete remission;
“CRO” means Contract Research Organization;
"CsA" means cyclosporine A;
"CSO" means Chief Scientific Officer;
“CTA” means clinical trial application;
"cyclosporine" means a drug that suppresses the immune system and is used to prevent rejection following organ transplantation;
"DES" means Dry Eye Syndrome;
"December 2016 Warrants" means the Common Share purchase warrants offered in connection with the US$28.75 million financing (including US$3.75 million pursuant to an exercise of the underwriters’ over-allotment option) for the sale of 12,778,000 units which closed on December 28, 2016.
"EDGAR" means the Electronic Data Gathering, Analysis and Retrieval System;
"eGFR" means estimated glomerular filtration rate;
"EMA" means the European Medicines Agency;
"ERA-EDTA" means the 54th European Renal Association-European Dialysis and Transplant Association Congress;
"ESRD" means end-stage renal disease;
"EU" means European Union;
"EULAR 2017" means the European Annual Congress of Rheumatology;
"Exchange Rules" means the rules of any stock exchange on which the Company's shares are listed for trading;
"FDA" means the Food and Drug Administration of the United States Government;
"FSGS" means focal segmental glomerulosclerosis;
"GMP" means good manufacturing practices;
"IEC" means Independent Ethics Committee;
"IFRS" means International Financial Reporting Standards;
"ILJIN" means ILJIN SNT Co., Ltd.;
"IND" means investigational new drug;
"IRB" means Institutional Review Board;
"ITT" means intent to treat;
"July 2016 ATM" means the at the market offering of Common Shares with an aggregate offer price of up to US$10 million;
"LN" means Lupus Nephritis;
"Lonza" means Lonza Ltd. a Swiss-based contract drug manufacturer;
"Lux" means Lux BioSciences, Inc.;
"March Offering" means the underwritten public offering of 25.64 million Common Shares, which included 3.35 million Common Shares issued pursuant to the full exercise of the underwriters’ overallotment option to purchase additional Common Shares;
"MMF" means mycophenolate mofetil;
"MPA" means mycophenolic acid, the active metabolite of MMF;
"MTT" means multi-targeted therapeutic;
"NASDAQ" means the NASDAQ Global Market Exchange;
"NCE" means new chemical entity;
"NDA" means New Drug Application made to a regulatory agency;
"Notice of Allowance" means the notice of allowance received from the USPTO for claims directed at our novel voclosporin dosing protocol for LN (U.S. patent application 15/835,219, entitled "PROTOCOL FOR TREATMENT OF LUPUS NEPHRITIS”);
"November 2016 ATM" means the at the market offering of Common Shares having an aggregate offer price of up to US$8.0 million;
"NS" means Nephrotic Syndrome;
"Paladin" means Paladin Labs Inc.;
"PCT" means the Patent Cooperation Treaty, an international patent law treaty. It provides a unified procedure for filing patent applications to protect inventions in each of its contracting states;
"Pharmacokinetics" means the processes of drug absorption, distribution, metabolism and escretion in a living system (e.g., in humans);
"PFIC" means a passive foreign investment company;
"PK-PD" means pharmacokinetic and pharmacodynamics analysis;
"PMDA" means the Pharmaceutical and Medical Devices Agency. The PMDA is the main Regulatory Agency that oversees the review and approval of drugs as per the regulatory prerequisites in Japan;
"PR" means partial remission;
"Registration Statement" means Aurinia's shelf registration statement on Form F-10 dated October 16, 2015, declared effective on November 5, 2015;
"SAE" means serious adverse events;
"SEC" means the U.S. Securities and Exchange Commission;
"SEDAR" means the System for Electronic Document Analysis and Retrieval;
"Sharp Clinical" means Sharp Clinical Services Inc.;
"SLE" means systemic lupus erythematosus;
"SLEDAI" means Systemic Lupus Erythematosus Disease Activity Index;
"SOC" means system organ class;
"TEAF" means treatment emergent adverse events;
"TSX" means the Toronto Stock Exchange;
"Unither" means Laboratoire Unither;
"UPCR" means Urinary/protein creatinine ratio;
"USPTO" means United States Patent and Trademark Office;
"Vifor" means Vifor (International) AG;
"VOS" means voclosporin ophthalmic solution; and
"Warrants" means warrants to purchase Common Shares in the capital of the Company, with each whole warrant being exercisable to purchase one common share.