Exhibit 99.2
Management's Discussion and Analysis

Third Quarter Ended September 30, 2020
MANAGEMENT’S DISCUSSION AND ANALYSIS OF
FINANCIAL CONDITION AND RESULTS OF OPERATIONS FOR
THE THIRD QUARTER ENDED SEPTEMBER 30, 2020
In this Management’s Discussion and Analysis of Financial Condition and Results of Operations ("MD&A"), unless the context otherwise requires, references to "we", "us", "our" or similar terms, as well as references to "Aurinia" or the "Company", refer to Aurinia Pharmaceuticals Inc., together with our subsidiaries.
The following MD&A provides information on the activities of Aurinia on a consolidated basis and should be read in conjunction with our unaudited interim condensed consolidated financial statements and accompanying notes for the three and nine months ended September 30, 2020 and our annual MD&A and audited financial statements for the year ended December 31, 2019. All amounts are expressed in United States (US) dollars unless otherwise stated. Dollar amounts in tabular columns are expressed in thousands of US dollars. This document is current in all material respects as of November 5, 2020.
The financial information contained in this MD&A and in our unaudited interim condensed consolidated financial statements has been prepared in accordance with International Financial Reporting Standards ("IFRS") as issued by the International Accounting Standards Board applicable to the preparation of interim financial statements including International Accounting Standard 34: Interim Financial Reporting. The unaudited interim condensed consolidated financial statements and MD&A have been reviewed and approved by our Audit Committee on November 5, 2020. This MD&A has been prepared with reference to National Instrument 51-102 "Continuous Disclosure Obligations" of the Canadian Securities Administrators. Under the US/Canada Multijurisdictional Disclosure System, Aurinia is permitted to prepare this MD&A in accordance with the disclosure requirements of Canada, which are different from those in the United States.
FORWARD-LOOKING STATEMENTS
A statement is forward-looking when it uses what we know and expect today to make a statement about the future. Forward-looking statements may include words such as “anticipate”, “believe”, “intend”, “expect”, “goal”, “may”, “outlook”, “plan”, “seek”, “project”, “should”, “strive”, “target”, “could”, “continue”, “potential” and “estimated”, or the negative of such terms or comparable terminology. You should not place undue reliance on the forward-looking statements, particularly those concerning anticipated events relating to the development, clinical trials, regulatory approval, and marketing of our products and the timing or magnitude of those events, as they are inherently risky and uncertain.
Securities laws encourage companies to disclose forward-looking information so that investors can get a better understanding of our future prospects and make informed investment decisions. These statements, made in this MD&A, may include, without limitation:
• | our belief that both the Phase 2b lupus nephritis ("LN") AURA- LV ("AURA") clinical trial and the single double-blind, randomized, placebo controlled Phase 3 clinical trial for voclosporin in the treatment of LN ("AURORA") had positive results; |
• | our plans to seek regulatory approval of voclosporin for the potential treatment of LN and other podocytopathies; |
• | our expectation that regulatory approval of voclosporin as a treatment for LN by the U.S. Food and Drug Administration (“FDA") is reasonably assured; |
• | our belief in the duration of patent exclusivity for voclosporin and that the patents owned by us are valid; |
• | our belief in receiving extensions to patent life based on certain events or classifications; |
• | our expectation that, upon regulatory approval, patent protection for voclosporin will be extended in the United States and certain other major markets, including Europe and Japan, until at least October 2027; |
• | our expectation to not receive any royalty revenue pursuant to the 3SBio license in the foreseeable future; |
• | our plans and expectations and the timing of commencement, enrollment, completion and release of results of clinical trials; |
• | our intention to demonstrate our belief that voclosporin possesses pharmacologic properties with the potential to demonstrate best-in-class differentiation with first-in-class status for the treatment of LN outside of Japan; |
• | our belief of the key potential benefits of voclosporin in the treatment of LN; |
• | our belief that voclosporin has the potential to improve near and long-term outcomes in LN when added to mycophenolate mofetil ("MMF"); |
• | our belief that voclosporin has the potential to address critical needs for LN by controlling active disease rapidly, lowering the overall steroid burden, and doing so with a convenient oral twice-daily treatment regimen; |
• | our expectation to receive "new chemical entity" exclusivity for voclosporin in certain countries, which provides this type of exclusivity for five years in the United States and up to ten years in Europe; |
• | our belief that the voclosporin modification of a single amino acid of the cyclosporine molecule may result in a more predictable pharmacokinetic and pharmacodynamics relationship, an increase in potency, an altered metabolic profile, and easier dosing without the need for therapeutic drug monitoring; |
• | our target launch date for voclosporin as a treatment for LN in the United States, if approved, in the first quarter of 2021; |
• | our belief in voclosporin being potentially a best-in-class calcineurin inhibitor ("CNI") (the cornerstone of therapy for the prevention of organ transplant rejection) with benefits over existing commercially available CNIs; |
• | our belief that voclosporin has further potential to be effectively used across a range of therapeutic autoimmune areas including proteinuric kidney diseases; |
• | the anticipated commercial potential of voclosporin for the treatment of LN; |
1
• | our plan to expand the voclosporin renal franchise with additional renal indications and the exploitation of voclosporin in novel formulations for treatment of autoimmune related disorders; |
• | our belief that voclosporin, in combination with MMF, has the potential to significantly improve renal response rates in LN versus current standard of care; |
• | our intention to use the net proceeds from financings for the stated purposes; |
• | our belief that we have sufficient cash resources to adequately fund our plans through the end of 2022; |
• | our ability to evaluate voclosporin in additional proteinuric kidney diseases; |
• | our plan to file a marketing authorization application with the European Medicines Agency ("EMA") by the second quarter of 2021; |
• | statements concerning the potential market for voclosporin; |
• | our belief that additional patents may be granted worldwide based on our filings under the Patent Cooperation Treaty ("PCT"); |
• | our belief that patents corresponding to United States Patent No. 10,286,036 issued to us covering dosing protocol, with corresponding FDA granted label, for voclosporin in LN, could be granted with similar claims in all major global pharmaceutical markets; |
• | our strategy to become a global commercial biopharmaceutical company; |
• | our expectation that pricing for voclosporin will be lower in Europe and Japan than in the United States driven by the specific country’s pricing and reimbursement processes and regulations; |
• | our plan to evaluate voclosporin in pediatric patients; |
• | our belief that the annualized pricing for voclosporin for LN could range between $45,000 and $90,000; |
• | the potential impact of COVID-19 on our business operations, nonclinical and clinical trials, regulatory timelines, supply chain, and potential commercialization; and |
• | a Prescription Drug User Fee Act ("PDUFA") target action date of January 22, 2021. |
Such statements reflect our current views with respect to future events and are subject to risks and uncertainties and are necessarily based on a number of estimates and assumptions that, while considered reasonable by management, as at the date of such statements, are inherently subject to significant business, economic, competitive, political, regulatory, legal, scientific and social uncertainties and contingencies, many of which, with respect to future events, are subject to change. The factors and assumptions used by management to develop such forward-looking statements include, but are not limited to:
• | the assumption that we will be able to obtain approval from regulatory agencies on executable development programs with parameters that are satisfactory to us; |
• | the assumption that recruitment to clinical trials will occur as projected; |
• | the assumption that we will successfully complete and enroll our clinical programs in compliance with good clinical practices on a timely basis and meet regulatory requirements for approval of marketing authorization applications and new drug approvals, as well as favourable product labeling; |
• | the assumption that the planned studies will achieve positive results; |
• | the assumptions regarding the costs and expenses associated with our clinical trials and commercialization of voclosporin including that the COVID-19 pandemic will not have a significant impact on the costs and expenses planned for our clinical trials and commercialization of voclosporin; |
• | the assumption that regulatory requirements and commitments will be maintained; |
• | the assumption that we will be able to meet Good Manufacturing Practice (“GMP”) standards and manufacture and secure a sufficient supply of voclosporin on a timely basis to successfully complete the development and commercialization of voclosporin; |
• | the assumptions on the market value for the LN program; |
• | the assumption that our patent portfolio is sufficient and valid; |
• | the assumption that we will be able to extend our patents to the fullest extent allowed by law, on terms most beneficial to us; |
• | the assumptions about future market activity; |
• | the assumption that there is a potential commercial value for other indications for voclosporin; |
• | the assumption that market data and reports reviewed by us are accurate; |
• | the assumptions on the burn rate of Aurinia’s cash for operations; |
• | the assumption that another company will not create a substantial competitive product for our LN business without violating our intellectual property rights or regulatory exclusivity periods; |
• | the assumption that our current good relationships with our suppliers, service providers and other third parties will be maintained; |
• | the assumption that we will be able to attract and retain a sufficient amount of skilled staff; and/or |
• | the assumptions relating to the capital required to fund operations through the end of 2022. |
It is important to know that:
• | actual results could be materially different from what we expect if known or unknown risks affect our business, or if our estimates or assumptions turn out to be inaccurate. As a result, we cannot guarantee that any forward-looking statement will materialize and, accordingly, you are cautioned not to place undue reliance on these forward-looking statements; and |
• | forward-looking statements do not take into account the effect that transactions or non-recurring or other special items announced or occurring after the statements are made may have on our business. For example, they do not include the effect of mergers, acquisitions, other business combinations or transactions, dispositions, sales of assets, asset write-downs or other charges announced or occurring after the forward-looking statements are made. The financial impact of such transactions and non-recurring and other special items can be complex and necessarily depend on the facts particular to each of them. Accordingly, the expected impact cannot be meaningfully described in the abstract or presented in the same manner as known risks affecting our business. |
2
The factors discussed below and other considerations discussed in the "Risks and Uncertainties" section of this MD&A could cause our actual results to differ significantly from those contained in any forward-looking statements.
Such forward-looking statements involve known and unknown risks, uncertainties, and other factors that may cause our actual results, performance, or achievements to differ materially from any assumptions, further results, performance or achievements expressed or implied by such forward-looking statements. Important factors that could cause such differences include, among other things, the following:
• | difficulties we may experience in completing the development, marketing and commercialization of voclosporin; |
• | unknown impact and difficulties imposed by the COVID-19 pandemic on our business operations including nonclinical and clinical and our supply chain; |
• | legislative, regulatory and commercial activities; |
• | the need for additional capital in the future to continue to fund our development programs and commercialization activities, and the effect of capital market conditions and other factors on capital availability; |
• | difficulties, delays, or failures we may experience in the conduct of and reporting of results of our clinical trials for voclosporin, including unfavourable results; |
• | difficulties in meeting GMP standards and the manufacturing and securing of a sufficient supply of voclosporin on a timely basis to successfully complete the development and commercialization of voclosporin; |
• | difficulties, delays or failures in obtaining necessary regulatory approvals, including for the commercialization of voclosporin; |
• | difficulties in gaining alignment among the key regulatory jurisdictions, FDA , EMA and Pharmaceutical and Medical Devices Agency, which may require further clinical activities; |
• | not being able to extend our patent portfolio for voclosporin; |
• | our patent portfolio not covering all of our proposed or contemplated uses of voclosporin; |
• | the uncertainty that the FDA will approve the use of voclosporin for LN and that the label for such use will follow the dosing protocol pursuant to US Patent No. 10,286,036 granted on May 4, 2019; |
• | the market for the LN business (or any other indication for voclosporin) may not be as we have estimated; |
• | insufficient acceptance of and demand for voclosporin; |
• | difficulties obtaining adequate reimbursements from third party payors; |
• | difficulties obtaining formulary acceptance; |
• | competitors may arise with similar products; |
• | product liability, patent infringement and other civil litigation; |
• | injunctions, court orders, regulatory and other compliance issues or enforcement actions; |
• | we may have to pay unanticipated expenses, and/or estimated costs for clinical trials or operations may be underestimated, resulting in our having to make additional expenditures to achieve our current goals; |
• | difficulties, restrictions, delays, or failures in obtaining appropriate reimbursement from payors for voclosporin; |
• | difficulties in retaining key personnel and attracting other qualified individuals; |
• | our assets or business activities may be subject to disputes that may result in litigation or other legal claims; |
• | difficulties we may experience in identifying and successfully securing appropriate vendors to support the development and commercialization of our product; |
• | our ability to raise future resources when required; and |
• | a change to the anticipated PDUFA target action date of January 22, 2021. |
Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance or achievements. These forward-looking statements are made as of the date hereof and we disclaim any intention and have no obligation or responsibility, except as required by law, to update or revise any forward-looking statements, whether as a result of new information, future events or otherwise.
For additional information on risks and uncertainties in respect of the Company and its business, please see the "Risks and Uncertainties" section of this MD&A. Although we believe that the expectations reflected in such forward-looking statements and information are reasonable, undue reliance should not be placed on forward-looking statements or information because we can give no assurance that such expectations will prove to be correct.
Additional information related to Aurinia, including its most recent Annual Information Form, is available by accessing the Canadian Securities Administrators’ System for Electronic Document Analysis and Retrieval ("SEDAR") website at www.sedar.com or the U.S. Securities and Exchange Commission’s Electronic Document Gathering and Retrieval System ("EDGAR") website at www.sec.gov/edgar.
OVERVIEW
THE COMPANY
Aurinia is a late-stage clinical biopharmaceutical company focused on developing and commercializing therapies to treat targeted patient populations that are suffering from serious diseases with a high unmet medical need. We are currently developing voclosporin, an investigational drug, for the potential treatment of LN and proteinuric kidney diseases.
3
Aurinia Pharmaceuticals Inc. is organized under the Business Corporations Act (Alberta). Our common shares (the "Common Shares") are currently listed and traded on the Nasdaq Global Market under the symbol "AUPH" and on the Toronto Stock Exchange ("TSX") under the symbol "AUP".
We have two wholly-owned subsidiaries: Aurinia Pharma U.S., Inc., (Delaware incorporated) and Aurinia Pharma Limited (United Kingdom incorporated).
Our head office is located at #1203-4464 Markham Street, Victoria, British Columbia, Canada and our registered office is located at #201, 17873 -106A Avenue, Edmonton, Alberta, Canada. Our US commercial office is located at 77 Upper Rock Circle, Rockville, Maryland.
BUSINESS OF THE COMPANY
Based in Victoria, British Columbia, Aurinia seeks to become a commercial-stage biopharmaceutical company. We are currently developing voclosporin, as a novel and potentially best-in-class CNI with clinical data in over 2,600 patients across various indications including LN, transplantation, psoriasis, various forms of uveitis, proteinuric kidney disease and dry eye syndrome ("DES").
On July 21, 2020, we announced that the FDA has accepted the filing of the new drug application ("NDA") for voclosporin, as a potential treatment for LN. The last element of this rolling NDA was submitted on May 26, 2020, after our December 4, 2019 release of positive AURORA Phase 3 trial results. The FDA has granted Priority Review for the NDA, which provides an expedited six-month review, and has assigned a PDUFA target action date of January 22, 2021. The FDA has also informed us that they are not currently planning to hold an advisory committee meeting to discuss the application. The FDA has the option to change this decision based on the review of the pending NDA. There are currently no FDA-approved treatments for LN.
Priority review is granted to therapies that the FDA determines have the potential to provide a significant improvement in the safety or effectiveness of the treatment, diagnosis or prevention of a serious condition. Under PDUFA, a Priority Review targets a review time of six months compared to the standard review time of 10 months. Voclosporin was also granted Fast Track designation by the FDA in 2016.
In addition, a marketing authorization application ("MAA") is planned to be filed with the EMA by the end of the second quarter of 2021.
Voclosporin is an immunosuppressant, with a synergistic and dual mechanism of action that has the potential to improve near and long-term outcomes in LN when added to MMF, the current standard of care for LN (although not currently approved for such use). By inhibiting calcineurin, voclosporin reduces cytokine activation and blocks interleukin IL-2 expression and T-cell mediated immune responses. Voclosporin also potentially stabilizes podocytes, which can protect against proteinuria. Voclosporin is made by a modification of a single amino acid of the cyclosporine molecule. This modification may result in a more predictable pharmacokinetic and pharmacodynamic relationship, an increase in potency, an altered metabolic profile, and easier dosing without the need for therapeutic drug monitoring. Clinical doses of voclosporin studied to date range from 13 - 70 mg administered orally twice a day ("BID"). The mechanism of action of voclosporin has been validated with certain first generation CNIs for the prevention of rejection in patients undergoing solid organ transplants and in several autoimmune indications, including uveitis, keratoconjunctivitis sicca, psoriasis, rheumatoid arthritis, and for LN in Japan. We believe that voclosporin possesses pharmacologic properties with the potential to demonstrate best-in-class differentiation with first-in-class regulatory approval status for the treatment of LN outside of Japan.
Legacy CNIs have demonstrated efficacy for a number of conditions, including transplant and other autoimmune diseases; however, side effects exist which can limit their long-term use and tolerability. Some clinical complications of legacy CNIs include hypertension, hyperlipidemia, diabetes, and both acute and chronic nephrotoxicity.
Based on published data, we believe the key potential benefits of voclosporin in the treatment of LN versus marketed CNIs are:
• | increased potency compared to cyclosporine A, allowing lower dosing requirements and potentially fewer off target effects; |
• | limited inter and intra patient variability, allowing for easier dosing without the need for therapeutic drug monitoring; |
• | less cholesterolemia and triglyceridemia than cyclosporine A; and |
• | limited incidence of glucose intolerance and diabetes at therapeutic doses compared to tacrolimus. |
Our target launch date for voclosporin as a treatment for LN in the United States, if approved, is the first quarter of 2021.
LN
LN is an inflammation of the kidney caused by systemic lupus erythematosus ("SLE") and represents a serious progression of SLE. SLE is a chronic, complex and often disabling disorder. The disease is highly heterogeneous, affecting a wide range of organs and tissue systems. Unlike SLE, LN has straightforward disease outcomes (measuring proteinuria) where an early response correlates with long-term outcomes. In patients with LN, renal damage results in proteinuria and/or hematuria and a decrease in renal function as evidenced by reduced estimated glomerular filtration rate ("eGFR"), and increased serum creatinine levels. eGFR is assessed through the Chronic Kidney Disease Epidemiology Collaboration equation. In 2004, a study indicated rapid control and reduction of proteinuria in LN patients measured at six months showed a reduction in the need for dialysis at 10 years. LN can be debilitating and costly and if poorly controlled, can lead to permanent and irreversible tissue damage within the kidney. Recent literature suggests LN can progress to end-stage renal disease ("ESRD") within 15 years of diagnosis in 10%-30% of patients, thus making LN a serious and potentially life-threatening condition. SLE patients with renal damage have a 14-fold increased risk of
4
premature death, while SLE patients with ESRD have a greater than 60-fold increased risk of premature death. In 2009, mean annual cost for patients (both direct and indirect) with SLE (with no nephritis) have been estimated to exceed $20,000 per year per patient, while the mean annual cost for patients (both direct and indirect) with LN who progress to intermittent ESRD have been estimated to exceed $60,000 per year per patient.
Phase 2/3 AUDREYTM Clinical Trial
On November 2, 2020 we announced topline data from the Phase 2/3 AUDREY™ clinical study evaluating voclosporin ophthalmic solution ("VOS") for the potential treatment of DES. The trial did not achieve statistical significance on its primary endpoint of a 10mm or greater improvement in STT at four weeks between active dose groups of VOS compared to vehicle. Aurinia is suspending the development program for VOS based upon these results.
The AUDREY™ trial was a randomized, double-masked, vehicle-controlled, dose-ranging study evaluating the efficacy and safety of VOS in subjects with DES. A total of 508 subjects were enrolled. The study consisted of four arms with a 1:1:1:1 randomization schedule, in which patients received either 0.2% VOS, 0.1% VOS, 0.05% VOS or vehicle, dosed twice daily for 12 weeks. The primary outcome measure for the trial was the proportion of subjects with a 10mm or greater improvement in STT at four weeks.
Secondary outcome measures evaluated in the trial included STT at other time points, Fluorescein Corneal Staining (FCS) at multiple time points, change in eye dryness, burning/stinging, itching, photophobia, eye pain and foreign body sensation at multiple time points, and additional safety endpoints. Initial analysis of these secondary outcomes suggests dose-dependent activity and safety were observed across dose groups compared to vehicle.
Investigator-Initiated Trial to Evaluate Antiviral Activity of Voclosporin in Kidney Transplant Recipients with COVID-19 ("VOCOVID")
On October 27, 2020 we announced the funding and initiation of an open-label exploratory trial evaluating the antiviral effects of voclosporin in kidney transplant recipients ("KTRs") with COVID-19 ("SARS-CoV-2") or the VOCOVID study. The single-center, investigator-initiated trial ("IIT") is being conducted by Drs. Aiko P.J. de Vries and Y.K. Onno Teng at the Leiden University Medical Center ("LUMC") in the Netherlands and will compare voclosporin against tacrolimus.
Organ transplant recipients who contract COVID-19 are at greater risk for complications due to the requirement of daily immunosuppressive medications to prevent organ rejection. CNIs, like voclosporin, have been shown in prior in vitro studies to inhibit viral replication. The team at the LUMC demonstrated that voclosporin inhibited viral replication of SARS-CoV-2 at an 8-fold lower concentration than tacrolimus in vitro, while maintaining cell viability of infected cells. In contrast to voclosporin, tacrolimus did not show antiviral activity against SARS-CoV-2 in vitro at clinically relevant concentrations. Therefore, given its potency and dosing advantages, voclosporin is a potentially attractive CNI for COVID-19 infected transplant patients who are already using legacy CNIs as part of their chronic immunosuppressive therapy.
This 56-day open-label investigator initiated trial (IIT) is designed to evaluate the antiviral effects of voclosporin compared to tacrolimus in stable kidney transplant recipients (KTRs) who contracted SARS-CoV-2. At study entry, 20 KTRs testing positive for SARS-CoV-2 and currently on dual immunosuppressives of prednisone and tacrolimus will be randomized 1:1 to remain on tacrolimus or be switched to voclosporin. The primary endpoint is the reduction in SARS-CoV-2 viral load over 56 days, as measured by reverse transcription polymerase chain reaction (qRT-PCR). The study will also assess predefined endpoints as surrogate markers of improved viral clearance including time to 3-log reduction in viral load concentration, time to clinical recovery – defined as free of symptoms for five days or more, and safety and tolerability. Following the 56-day treatment period, there will be an extended safety follow-up of voclosporin treated patients for up to one year.
Market Potential and Commercial Considerations
We have conducted market research including claims database reviews (where available) and physician-based research. Our physician research included approximately 900 rheumatologists and nephrologists across the United States, Europe and Japan to better define the potential market size, estimated pricing and treatment paradigms in those jurisdictions. In an updated review of the Symphony Integrated Dataverse (IDV®) claims database from 2017 using ICD-10 SLE diagnosis codes, there were 421,790 individuals in that database. The National Institute of Diabetes and Digestive and Kidney Diseases estimates that up to 50% of adults with SLE are diagnosed with kidney disease at some point in their journey with lupus. Using the latest claims database research, we estimate the number of SLE patients diagnosed with kidney involvement to be no more than 150,000 in the United States and 150,000 to 215,000 for Europe and Japan combined. Similar to other autoimmune disorders, LN is a flaring and remitting disease. The destructive disease cycle people with LN go through is depicted below. The disease cycles from being in remission to being in flare, achieving partial remission and being back in remission. Treatment objectives between LN and other autoimmune diseases are remarkably similar. In other autoimmune conditions such as Multiple Sclerosis, Crohn’s, Rheumatoid Arthritis and SLE, physicians’ goals are to induce/maintain a remission of disease, decrease frequency of hospital or ambulatory care visits and limit long term disability. In LN specifically, physicians are trying to avoid further kidney damage, dialysis, renal transplantation, and death. According to a physician survey, the frequency of LN flares amongst treated patients was approximately every 14 months across the United States and Europe. The ability to get patients into remission quickly correlates with better long-term kidney outcomes as noted above.
5
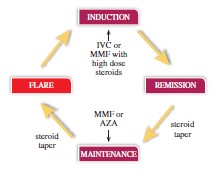
The population of people with LN will be in different cycles of their disease at any one time. Physicians currently use existing LN standard of care including immunosuppressants and high dose steroids to treat people with LN throughout the disease cycles including induction and maintenance phases. By studying voclosporin on top of an existing standard of care we are not seeking to displace current accepted treatment patterns. We believe that the potential to be additive to an existing standard of care in addition to voclosporin being administered orally, rather than via infusion or injection, can support a more rapid market adoption if approved. Current annualized pricing (based on wholesale acquisition costs published by AnalySource®, reprinted with permission by First Databank, Inc.) for the treatments of other more prevalent autoimmune conditions such as Crohn’s, Rheumatoid Arthritis and SLE ranges from $45,000 to $90,000 in the United States. Of course, pricing is highly variable and dependent on a wide variety of factors, including the cost of manufacturing the product, the value perceived by physicians, regulatory concerns, payor policies, and political landscape, along with other market factors that may exist at the time the product is ready to be marketed. Wholesale acquisition cost is the manufacturer’s published catalog or list price for a drug product to wholesalers and may not reflect actual prices paid after any rebates/discounts. We have conducted preliminary pricing research that studied a similar pricing range with payors and physicians and believe that pricing in this range may be achievable for voclosporin in the United States. Pricing for other autoimmune conditions is lower in Europe and Japan than it is in the United States, which is driven by country specific pricing and reimbursement processes. We expect that will be the case for voclosporin.
STRATEGY
Our business strategy is to optimize the clinical and commercial value of voclosporin and become a global commercial biopharmaceutical company with a focused renal and autoimmune franchise. This includes the expansion of a potential renal franchise with additional renal indications and the exploitation of voclosporin in novel formulations for the treatment of autoimmune related disorders.
We have developed a plan to expand our voclosporin renal franchise to include proteinuric kidney diseases beyond LN.
The key tactics to achieve our corporate strategy include:
• | filing an NDA with the FDA for marketing approval for use of voclosporin in LN by the end of the second quarter of 2020, which was completed on May 26, 2020; |
• | conducting pre-commercial activities including build out of the organization to efficiently and effectively launch voclosporin as a treatment for LN upon potential approval by the FDA; |
• | evaluating voclosporin in additional proteinuric kidney diseases; and |
• | evaluating assets with the potential to be synergistic and complementary to our clinical, regulatory and therapeutic area of expertise. |
CORPORATE DEVELOPMENTS IN Q3 2020
July 27, 2020 Public Offering
On July 27, 2020 we completed an underwritten public offering of 13.33 million Common Shares (the “July 2020 Offering”).
The Common Shares were sold at a public offering price of $15.00 per share. The gross proceeds from the July 2020 Offering were $200 million before deducting the 6% underwriting commission and other offering expenses which totaled an estimated aggregate $12.50 million. Jefferies and SVB Leerink acted as joint book-running managers for the July 2020 Offering. Cantor acted as lead manager and Oppenheimer & Co and H.C. Wainwright & Co. acted as co-managers for the July 2020 Offering. We intend to use the net proceeds of the July 2020 Offering for pre-commercialization and launch activities, research and development ("R&D"), as well as working capital and general corporate purposes.
Submission of NDA to the FDA
On July 21, 2020, we announced that FDA has accepted the filing of the NDA for voclosporin, as a potential treatment for LN. The FDA has granted Priority Review for the NDA, which provides an expedited six-month review, and has assigned a PDUFA target action date of January 22, 2021. The FDA has also informed us that they are not currently planning to hold an advisory committee meeting to discuss the application. The FDA has the option to change this decision based on the review of the pending NDA.
6
Priority review is granted to therapies that the FDA determines have the potential to provide a significant improvement in the safety or effectiveness of the treatment, diagnosis or prevention of a serious condition. Under PDUFA, a Priority Review targets a review time of six months compared to a standard review time of 10 months. Voclosporin was also granted Fast Track designation by the FDA in 2016.
There are currently no FDA-approved treatments for LN.
Investigator-Initiated Trial to Evaluate Antiviral Activity of Voclosporin in Kidney Transplant Recipients with COVID-19 ("VOCOVID")
On October 27, 2020 we announced the funding and initiation of an open-label exploratory trial evaluating the antiviral effects of voclosporin in kidney transplant recipients ("KTRs") with COVID-19 ("SARS-CoV-2") or the VOCOVID study. The single-center, investigator-initiated trial ("IIT") is being conducted by Drs. Aiko P.J. de Vries and Y.K. Onno Teng at the Leiden University Medical Center ("LUMC") in the Netherlands and will compare voclosporin against tacrolimus.
Organ transplant recipients who contract COVID-19 are at greater risk for complications due to the requirement of daily immunosuppressive medications to prevent organ rejection. CNIs, like voclosporin, have been shown in prior in vitro studies to inhibit viral replication. The team at the LUMC demonstrated that voclosporin inhibited viral replication of SARS-CoV-2 at an 8-fold lower concentration than tacrolimus in vitro, while maintaining cell viability of infected cells. In contrast to voclosporin, tacrolimus did not show antiviral activity against SARS-CoV-2 in vitro at clinically relevant concentrations. Therefore, given its potency and dosing advantages, voclosporin is a potentially attractive CNI for COVID-19 infected transplant patients who are already using legacy CNIs as part of their chronic immunosuppressive therapy.
This 56-day open-label investigator initiated trial (IIT) is designed to evaluate the antiviral effects of voclosporin compared to tacrolimus in stable kidney transplant recipients (KTRs) who contracted SARS-CoV-2. At study entry, 20 KTRs testing positive for SARS-CoV-2 and currently on dual immunosuppressives of prednisone and tacrolimus will be randomized 1:1 to remain on tacrolimus or be switched to voclosporin. The primary endpoint is the reduction in SARS-CoV-2 viral load over 56 days, as measured by reverse transcription polymerase chain reaction (qRT-PCR). The study will also assess predefined endpoints as surrogate markers of improved viral clearance including time to 3-log reduction in viral load concentration, time to clinical recovery – defined as free of symptoms for five days or more, and safety and tolerability. Following the 56-day treatment period, there will be an extended safety follow-up of voclosporin treated patients for up to one year.
Appointment of New Officer
On November 2, 2020 we announced the appointment of Stephen Robertson as Executive Vice President, General Counsel, Corporate Secretary and Chief Compliance Officer. He replaces Dr. Erik Eglite, who is leaving the Company to pursue other interests. Mr. Robertson will be responsible for all legal matters related to Aurinia, its investigational drug voclosporin and potential future in-licensing and out-licensing agreements.
Mr. Robertson brings more than 13 years of corporate law experience across various roles with the law firm Borden Ladner Gervais LLP, where he has been a Partner since 2014. He has focused on advising clients on securities, corporate and commercial legal matters, including extensive experience with mergers and acquisitions and commercial agreements. Mr. Robertson has served as Corporate Secretary for Aurinia since 2014.
Mr. Robertson received his Bachelor of Laws degree from the University of Manitoba. He has been recognized with a number of awards and honors, including being included in the 2020 edition of the Best Lawyers in Canada for Securities Law.
Upcoming Changes in IFRS / Foreign Private Issuer Status
We are a foreign private issuer (“FPI”) as defined under the U.S. Securities Exchange Act of 1934 (“Exchange Act”) and we utilize the multijurisdictional disclosure system (“MJDS”) as permitted for Canadian corporations for filing reports with the U.S. Securities and Exchange Commission (“SEC”). We are required under applicable rules to test our FPI status annually at the end of our second fiscal quarter. If an issuer fails to qualify as an FPI at the end of its second fiscal quarter, it remains eligible to use the forms and rules applicable to FPIs until the end of that financial year. Historically, we met the definition of an FPI, and as such, prepared our consolidated financial statements in accordance with IFRS and complied with SEC rules and regulations applicable to Canadian corporations filing reports using MJDS.
As of June 30, 2020, since more than 50% of our Common Shares are held by U.S. residents and a majority of our executive officers are U.S. citizens or residents, we no longer qualify as an FPI. Therefore, we will transition to U.S. domestic corporation reporting status and become subject to the applicable SEC reporting requirements beginning on January 1, 2021. These reporting requirements will require that our financial statements be presented in accordance with U.S. Generally Accepted Accounting Principles for all periods, which will include fiscal 2020 comparative financial information. Therefore, the last period under which we will report under IFRS is the third quarter ended September 30, 2020. In addition, we will be required to file annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K with the SEC, our executive officers and directors will be required to comply with Section 16 under the Exchange Act with respect to reporting transactions in our Common Shares, and we will be subject to the SEC’s proxy solicitation rules.
7
RESULTS OF OPERATIONS
For the three months ended September 30, 2020 we reported a consolidated net loss of $34.06 million or $0.28 loss per Common Share, as compared to a consolidated net loss of $19.04 million or $0.21 loss per Common Share for the three months ended September 30, 2019.
On a year-to-date basis, we recorded a consolidated net loss of $80.12 million or $0.69 per share for the nine months ended September 30, 2020, compared to a consolidated net loss of $47.37 million or $0.52 per share for the nine months ended September 30, 2019.
We recorded an increase in the estimated fair value of derivative warrant liabilities of $9.49 million for the nine months ended September 30, 2020 compared to recording $6.86 million for the nine months ended September 30, 2019.
After adjusting for the non-cash impact of the revaluation of the warrant liabilities, the net loss before change in estimated fair value of derivative warrant liabilities and income tax expense (recovery) for the three and nine months ended September 30, 2020 was $36.70 million and $89.87 million compared to $23.52 million and $54.17 million for the same period in 2019. The higher net loss before the change in estimated fair value of derivative warrant liabilities and income tax expense (recovery) in 2020 was primarily due to increases in corporate, administration and business development expenses for the nine months ended September 30, 2020.
Research and development expenses
R&D expenses decreased to $4.80 million and $29.71 million respectively for the three and nine month periods ended September 30, 2020 compared to $17.79 million and $39.57 million respectively for the three and nine month periods ended September 30, 2019. The decrease in R&D expenses for the nine months ended September 30, 2020 primarily reflected the decrease in activities related to clinical trials and exploratory development work. Additionally, in the three month period ended September 30, 2020 as management believes that the approval by the FDA of voclosporin as a treatment for LN is reasonably assured, the capitalization of inventory $6.29 million and internal development costs $1.18 million further decreased R&D expenses. There were no similar events in the comparable period.
Contract Research Organizations ("CROs") and other third party expenses were $4.79 million and $18.14 million respectively for the three and nine month periods ended September 30, 2020 compared to $6.80 million and $20.87 million respectively for the three and nine month periods ended September 30, 2019. The decrease in these costs reflected no AURORA trial costs partially offset by higher NDA submission preparation costs, higher CRO costs related to the now completed VOS Phase 2/3 dry eye trial and the ongoing AURORA 2 extension trial during the period.
We incurred drug manufacturing and supply recovery of $3.82 million and expense of $557,000 respectively for the three and nine month periods ended September 30, 2020, compared to $8.68 million and $11.36 million respectively for the three and nine month periods ended September 30, 2019. The decrease in these expenses primarily reflects the costs incurred related to the ongoing preparation of the chemistry, manufacturing and controls section of the NDA submission and commercial drug supply activities required for launch offset by the capitalization of previously expensed inventory and API costs of $6.29 million. Further information related to the inventory is more fully discussed in note 6 of the unaudited interim condensed consolidated financial statements for the three and nine months ended September 30, 2020.
Salaries, annual incentive pay accruals and employee benefits (excluding non-cash stock compensation expense noted below) increased to $2.65 million and $7.03 million respectively for the three and nine month periods ended September 30, 2020 compared to $1.41 million and $3.95 million respectively for the three and nine month periods ended September 30, 2019. The increase primarily reflected the hiring of additional R&D employees since January 1, 2020, annual salary increases effective January 1, 2020 and enhanced employee benefits over the past year.
Included in the R&D expenses was non-cash stock compensation expense of $814,000 and $3.11 million respectively for the three and nine month periods ended September 30, 2020 compared to $596,000 and $2.21 million respectively for the three and nine month periods ended September 30, 2019 for stock options granted to R&D personnel. See the section on stock-based compensation expense below for further details.
Other expenses, which included items such as travel, clinical trial insurance, patent annuity and legal fees, phone and publications were $367,000 and $880,000 for the three and nine month periods ended September 30, 2020 compared to $301,000 and $1.18 million for the three and nine month periods ended September 30, 2019. The decrease in costs primarily reflected less travel activity due to the COVID-19 pandemic in the first nine months of 2020.
Corporate, administration and business development expenses
Corporate, administration and business development expenses increased to $31.07 million and $57.67 million respectively for the three and nine month periods ended September 30, 2020 compared to $6.06 million and $14.91 million respectively for the three and nine month periods ended September 30, 2019 and reflect the investment incurred to build out our organization to support the launch of voclosporin as a treatment for LN which is planned for early 2021, subject to FDA regulatory approval being granted. Since the release of the positive results of our AURORA trial in December of 2019 we have moved quickly to develop our commercial capabilities across the organization including the expansion of the commercial team headed by our new Chief Commercial Officer.
Salaries, director fees, payroll accruals and employee benefits (excluding stock compensation expense noted below) were $13.92 million and $22.75 million for the three and nine month periods ended September 30, 2020 compared to $1.55 million and $4.54 million for the three and nine month periods ended September 30, 2019. The increases primarily reflected the hiring of 136 additional sales and commercial employees to support the commercialization of voclosporin during the three months ended September 30, 2020, compared to one new employee in the comparable period.
8
Corporate, administration and business development expenses included non-cash stock-based compensation expense of $3.75 million and $9.15 million for the three and nine month periods ended September 30, 2020 compared to $1.43 million and $3.38 million for three and nine month periods ended September 30, 2019. See the section on stock-based compensation expense below for further details.
Professional and consulting fees were $8.80 million and $18.20 million for the three and nine month periods ended September 30, 2020 compared to $1.95 million and $3.94 million for the three and nine month periods ended September 30, 2019. Higher professional and consulting fees were incurred in 2020 related to recruiting, legal, tax advice, human resources, information technology and pre-commercialization activities including market research, market access, patience advocacy and communications. The increase reflected significant expansion in activity levels across the organization during the three and nine months ended September 30, 2020.
Rent, insurance, information technology, communications and other public company operating costs increased to $4.0 million and $6.43 million for the three and nine month periods ended September 30, 2020 compared to $631,000 and $1.66 million for the three and nine month periods ended September 30, 2019. The increase reflected overall higher activity levels, higher staff numbers, and higher director and officer insurance costs commensurate with the Company's growth and preparations to support the commercialization of voclosporin.
Travel, tradeshows and sponsorships increased to $603,000 for the three month period ended September 30, 2020 compared to $501,000 for the three month period ended September 30, 2019. This was due to our increased participation in virtual conferences and tradeshows. Travel, tradeshows and sponsorships decreased to $1.14 million for the nine month period ended September 30, 2020 compared to $1.39 million for the nine month period ended September 30, 2019 reflecting higher activity levels in 2019 combined with the evolving opportunity to participate in conferences and tradeshows virtually, as a result of the COVID-19 pandemic.
Other expenses
Other expenses were $426,000 and $2.35 million for the three and nine month periods ended September 30, 2020 compared to $140,000 and $1.03 million for the three and nine month periods ended September 30, 2019.
Other expenses were comprised of the following:
Royalty Obligation expense
The royalty obligation is the result of a resolution of the Board of Directors of the Company dated March 8, 2012 whereby certain executive officers at that time were provided with future potential retention benefits for remaining with the Company as follows:
(a) Pursuant to a resolution of the Board of Directors of the Company on March 8, 2012 and a termination agreement and general release dated February 14, 2014, the Company will be required to pay a royalty, equivalent to 2% of royalties received on the sale of voclosporin by licensees and/or 0.3% of net sales of voclosporin sold directly by the Company to the Chief Executive Officer at the time of the resolution. Should the Company sell substantially all of the assets of voclosporin to a third party or transfer those assets to another party in a merger in a manner such that this payment obligation is no longer operative, then the Company would be required to pay 0.3% of the value attributable to voclosporin in the transaction.
(b) In addition, pursuant to a resolution of the Board of Directors of the Company on March 8, 2012, and employment agreements, two other executive officers of the Company at the time of the resolution are eligible to receive 0.1675% of royalty licensing revenue for royalties received on the sale of voclosporin by licensees and/or 0.025% of net sales of voclosporin sold directly by the Company. Should the Company sell substantially all of the assets of voclosporin to a third party or transfer those assets to another party in a merger, the executives will be entitled to receive 0.025% of the value attributable to voclosporin in the transaction, and the entitlement to further royalty or sales payments shall end. This entitlement is terminated upon the death of the individual.
The Board of Directors resolution, dated March 8, 2012, created an employee benefit obligation contingent on the occurrence of uncertain future events. The probability that the specified events will occur affects the measurement of the obligation.
As a result of the completion and results obtained of the AURORA trial, we re-assessed the probability of royalty obligation payments being required in the future, and have recorded the royalty obligation at December 31, 2019. Until one of the triggering events in sections (a) or (b) described above occur, no royalty payments are required to be paid. No material royalties on sales or licensing are expected to be paid in the next twelve months and therefore the royalty obligation has been classified as long term. The fair value of the royalty obligation as at September 30, 2020 was estimated to be $8.00 million (December 31, 2019 - $7.20 million).
During the three months ended September 30, 2020 we re-assessed the royalty obligation and increased the discount rate to 11% as of September 30, 2020, compared to 10.5% at June 30, 2020. The change in discount rate, increased probability and the passage of time, on revaluation, resulted in a royalty obligation increase of $300,000. For the nine month period ended September 30, 2020 there was an increase of $800,000 in the royalty obligation which was primarily attributable to the increased probability, passage of time, on revaluation, combined with the decrease of discount rate from 12% at December 31, 2019 to 11% at September 30, 2020. There were no similar adjustments for the three and nine month periods ended September 30, 2019.
Revaluation adjustment on contingent consideration
The fair value estimates at September 30, 2020 were based on a weighted average discount rate of 1.6% (December 31, 2019 - 10%) and a presumed payment range between 50% and 90% (December 31, 2019 - 50% and 86%). The decrease of the discount rate was primarily attributable
9
to the significant decline in interest rates caused by the COVID-19 pandemic. The fair value of this contingent consideration as at September 30, 2020 was estimated to be $6.33 million (December 31, 2019 - $5.11 million) and was determined by estimating the probability and timing of achieving the milestones and applying the income approach. The change in discount rate, probability, and passage of time, on revaluation, resulted in an increase in contingent consideration of $352,000 and $1.22 million respectively for the three and nine months ended September 30, 2020 compared to an increase in contingent consideration of $127,000 and $207,000 respectively for the three and nine months ended September 30, 2019.
Foreign exchange loss
We incurred a foreign exchange gain of $226,000 and a foreign exchange loss of $332,000 respectively for the three and nine month periods ended September 30, 2020 compared to a foreign exchange loss of $13,000 and $101,000 for the comparable period in 2019. The increase in this loss for the nine months ended September 30, 2020 was primarily the result of a significant decrease in the Canadian dollar against the U.S. dollar at March 31, 2020, compared to December 31, 2019 which resulted in a loss of $1.02 million, combined with an increase in the Canadian dollar against the U.S. dollar at September 30, 2020 compared to March 31, 2020. The majority of this loss is unrealized and in the future will result either in lower costs for our Canadian operations when converted to U.S. dollars or be reversed if the Canadian dollar strengthens against the U.S. dollar. Our position in Canadian dollars is a result of the exercise of stock options which are denominated in Canadian dollars.
Interest Income
We recorded interest income of $170,000 and $1.38 million for the three and nine month periods ended September 30, 2020 compared to $636,000 and $2.23 million for the three and nine month periods ended September 30, 2019 reflecting a decrease in interest rates due to COVID offset by our higher cash position.
Stock-based compensation expense
For stock option plan information and outstanding stock option details refer to note 11(b) of the unaudited interim condensed consolidated financial statements for the three and nine months ended September 30, 2020.
We granted 967,000 and 5.15 million stock options for the three and nine month periods ended September 30, 2020 at a weighted average exercise price of $14.91 and $16.78 respectively compared to 645,000 and 3.73 million stock options at a weighted average exercise price of $6.02 and $6.16 for the three and nine month periods ended September 30, 2019. The grants noted above included nil and 1.37 million stock options granted to executive officers for the three and nine month periods ended September 30, 2020 at a weighted average exercise price of $nil and $17.71 respectively compared to 250,000 and 2.73 million granted at a weighted average exercise price of $5.90 and $6.23 for the same period in 2019. Of the stock options issued in the three month period ended September 30, 2020, 530,000 were inducement stock options to 105 new employees entering into employment with the Company in accordance with Nasdaq listing rule 5635(c)(4) at a price of $14.83 (CA$19.37). For the purposes of TSX approval, the Company relied on the exemption set forth in Section 602.1 of the TSX Company Manual, which provides that the TSX will not apply its standards to certain transactions involving eligible inter-listed issuers on a recognized exchange.
Application of the fair value method resulted in charges to stock-based compensation expense of $4.61 million and $12.31 million for the three and nine month periods ended September 30, 2020 (2019 – $2.02 million and $5.59 million) with corresponding credits to contributed surplus. For the three and nine month periods ended September 30, 2020, R&D stock compensation expense was $814,000 and $3.11 million (September 30, 2019 – $596,000 and $2.21 million) and corporate, administration and business development stock compensation expense was $3.75 million and $9.15 million (September 30, 2019 – $1.43 million and $3.38 million).
Stock compensation expense related to executive officers was $2.22 million and $6.31 million respectively for the three and nine months ended September 30, 2020 compared to $1.39 million and $3.52 million respectively for the three and nine months ended September 30, 2019.
The increase in stock option compensation expense for the nine months ended September 30, 2020 reflected higher stock option grants resulting from the hiring of 199 new employees since January 1, 2020 and an increase in the fair value of the stock options granted due to the significant increase in our share price.
Amortization of acquired intellectual property and other intangible assets
Amortization of acquired intellectual property and other intangible assets was $348,000 and $1.04 million for the three and nine month periods ended September 30, 2020 compared to $348,000 and $1.04 million for the same periods in 2019.
Change in estimated fair value of derivative warrant liabilities
We recorded a non-cash decrease in estimated fair value of derivative warrant liabilities of $2.60 million and non-cash decrease in estimated fair value of derivative warrant liabilities of $9.49 million respectively for the three and nine month periods ended September 30, 2020 compared to a non-cash decrease of $4.51 million and $6.86 million for the three and nine month periods ended September 30, 2019. These revaluations fluctuate based primarily on the market price of our Common Shares. Derivative warrant liabilities are more fully discussed in the section Critical estimates in applying the Company’s accounting policies and note 10 (Derivative Warrant Liabilities) of the unaudited interim condensed consolidated financial statements for the three and nine months ended September 30, 2020.
10
In accordance with IFRS, a contract to issue a variable number of shares fails to meet the definition of equity and must instead be classified as a derivative liability and measured at fair value with changes in fair value recognized in the consolidated statements of operations and comprehensive loss at each period-end. To clarify, while we will settle these warrants only in shares in the future, accounting rules require that we show a liability because of the potential variability in the number of shares which may be issued if the cashless exercise option is used by the holder of the warrants under the specific situations discussed below.
The derivative warrant liabilities will ultimately be eliminated on the exercise or forfeiture of the warrants and will not result in any cash outlay by the Company.
Derivative warrant liability related to December 31, 2016 bought deal public offering
On December 28, 2016, we completed a $28.75 million bought deal public offering (the "December Offering"). Under the terms of the December Offering, we issued 12.78 million units at a subscription price per unit of $2.25, each unit consisting of one Common Share and one-half (0.50) of a Common Share purchase warrant (a "Warrant"), exercisable for a period of five years from the date of issuance at an exercise price of $3.00. Therefore, we issued 6.39 million Warrants. The holders of the Warrants issued pursuant to the December Offering may elect, if we do not have an effective registration statement registering the Common Shares underlying the Warrants, or the prospectus contained therein is not available for the issuance of the Common Shares underlying the Warrants to the holder, in lieu of exercising the Warrants for cash, a cashless exercise option to receive Common Shares equal to the fair value of the Warrants. This calculation is based on the number of Warrants to be exercised multiplied by the weighted average market price less the exercise price with the difference divided by the weighted average market price. If a Warrant holder exercises this option, there will be variability in the number of shares issued per Warrant. There can be no certainty that we will have an effective registration statement in place over the entire life of the Warrants and therefore, under IFRS we are required to record these Warrants as derivative warrant liabilities. These Warrants will expire on December 28, 2021.
There were no derivative warrant exercises during the three months ended September 30, 2020. During the three months ended March 31, 2020, a holder exercised 500 Warrants for $3.00 per share, for gross proceeds of $1,500. There were no derivative warrant exercises for the three or nine month periods ended September 30, 2019 related to these warrants.
At September 30, 2020, there were 1.69 million of the December 28, 2016 Warrants outstanding at an exercise price of $3.00.
LIQUIDITY AND CAPITAL RESOURCES
At September 30, 2020, we had cash and cash equivalents and short term investments on hand of $392.04 million compared to cash and cash equivalents of $306.02 million at December 31, 2019.
We are a development stage company and are devoting the majority of our operational efforts and financial resources towards the clinical development and potential commercialization of our late stage drug, voclosporin. For the nine months ended September 30, 2020, we reported a loss of $80.12 million (September 30, 2019 - $47.37 million) and a cash outflow from operating activities of $75.32 million (September 30, 2019 - $38.18 million). As of September 30, 2020, we had an accumulated deficit of $619.93 million (September 30, 2019 - $463.33 million).
We believe that our cash position is sufficient to fund our current plans which include conducting our planned R&D programs, funding pre-commercial and launch activities, manufacturing and packaging of commercial drug supply required for launch, and funding our supporting corporate and working capital needs through the end of 2022.
Sources and Uses of Cash:
Three months ended September 30, | Nine months ended September 30, | ||||||||
2020 (in thousands) | 2019 (in thousands) | 2020 (in thousands) | 2019 (in thousands) | ||||||
$ | $ | $ | $ | ||||||
Cash used in operating activities | (30,280 | ) | (11,775 | ) | (75,315 | ) | (38,177 | ) | |
Cash (used in) generated from investing activities | (142,633 | ) | (30 | ) | (175,079 | ) | 7,812 | ||
Cash generated from financing activities | 189,257 | 14,851 | 193,133 | 46,937 | |||||
Net increase (decrease) in cash and cash equivalents | 16,344 | 3,046 | (57,261 | ) | 16,572 | ||||
Cash used in operating activities was $30.28 million and $75.32 million respectively for the three and nine month periods ended September 30, 2020 compared to cash used in operating activities of $11.78 million and $38.18 million for the same periods in 2019. Cash used in operating activities was composed of net loss, add-backs or adjustments not involving cash, such as stock-based compensation, reversal of inventory provision, royalty obligation, and change in estimated fair value of derivative warrant liabilities and net change in other operating assets and liabilities including prepaid expenses and deposits and accounts payable and accrued liabilities.
Cash used in investing activities was $142.63 million and $175.08 million respectively for the three and nine month periods ended September 30, 2020 compared to cash used in investing activities of $30,000 and cash generated in investing activities of $7.81 million respectively for
11
the same periods in 2019. The change in these amounts primarily related to movements within our investment portfolio and purchases of property, plant and equipment.
Cash generated from financing activities was $189.26 million and $193.13 million respectively for the three and nine month periods ended September 30, 2020 compared to cash generated by financing activities of $14.85 million and $46.94 million for the same periods in 2019. Cash generated from financing activities for the three and nine month periods ended September 30, 2020 was primarily from the issuance of shares pursuant to the July 2020 Offering (as defined below) which resulted in net proceeds of $187.73 million. Cash generated from financing activities for the nine month period ended September 30, 2019 was primarily comprised of net proceeds of $14.28 million from the 2019 ATM (as defined below).
Use of Financing Proceeds
March 2017 Offering
On March 20, 2017, we completed an underwritten public offering of 25.64 million Common Shares, which included 3.35 million Common Shares issued pursuant to the full exercise of the underwriters' overallotment option to purchase additional Common Shares, for net proceeds of $162.32 million, which are to be used for R&D activities and for working capital and corporate purposes.
November 2018 ATM
On November 30, 2018 we entered into an open market sale agreement with Jefferies LLC pursuant to which Aurinia would be able to, from time to time, sell, through at-the-market ("ATM") offerings, Common Shares that would have an aggregate offering price of up to $30.00 million (the "2018 ATM"). As of the first quarter of 2019, the agreement terminated as the maximum dollar amount of Common Shares were sold under the 2018 ATM. We received net proceeds of $28.83 million from the 2018 ATM. The net proceeds are to be used for working capital and corporate purposes.
September 2019 ATM
On September 13, 2019 we entered into an open market sale agreement with Jefferies LLC pursuant to which Aurinia would be able to, from time to time, sell, through ATM offerings, Common Shares that would have an aggregate offering price of up to $40.00 million (the "2019 ATM"). On December 9, 2019 we terminated the agreement with Jefferies LLC related to the 2019 ATM. We received net proceeds of $14.37 million from the 2019 ATM. The net proceeds are to be used for working capital and corporate purposes.
December 2019 Offering
On December 12, 2019, we completed an underwritten public offering of 12.78 million Common Shares, which included 1.67 million Common Shares issued pursuant to the full exercise of the underwriters’ overallotment option to purchase additional Common Shares, for net proceeds of $179.92 million (the "December 2019 Offering"), which are to be used for pre-commercialization and launch activities, working capital and general corporate purposes.
July 2020 Offering
On July 27, 2020, we completed an underwritten public offering of 13.33 million Common Shares, for net proceeds of $187.73 million. The net proceeds will be used for pre-commercialization and launch activities, R&D activities, working capital and general corporate purposes.
12
A summary of the anticipated and actual use of net proceeds used to date from the above financings is set out in the table below:
Allocation of net proceeds | Total net proceeds from financings (in thousands) | Net proceeds used to date (in thousands) | ||
$ | $ | |||
March 2017 Offering: | ||||
R&D activities | 123,400 | 119,980 | ||
Working capital and corporate purposes | 38,924 | 38,924 | ||
Subtotal: | 162,324 | 158,904 | ||
November 2018 ATM facility | 28,830 | 28,830 | ||
September 2019 ATM facility | 14,371 | 8,206 | ||
December 2019 Public Offering: | ||||
Pre-commercial and launch activities, working capital and corporate purposes | 179,918 | — | ||
July 2020 Public Offering: | ||||
Pre-commercial and launch related activities | $117,000 to $143,000 | — | ||
R&D activities | $28,000 to $34,000 | — | ||
Subtotal: | 187,500 | — | ||
Total: | 572,943 | 195,940 | ||
As of September 30, 2020, there have been no material variances from how we disclosed we were going to use the proceeds from the above noted offerings and thus no material impact on its ability to achieve our business objectives and milestones.
CONTRACTUAL OBLIGATIONS
We have the following contractual obligations as at September 30, 2020:
Total (in thousands) | Less than one year (in thousands) | One to three years (in thousands) | Four to five years (in thousands) | More than five years (in thousands) | ||||||||||
$ | $ | $ | $ | $ | ||||||||||
Lease Liabilities (net of lease inducements) (1) | 11,339 | (372 | ) | 431 | 2,141 | 9,139 | ||||||||
Operating lease obligations (2) | 88 | 88 | — | — | — | |||||||||
Purchase obligations (3) | 7,674 | 7,614 | 60 | — | — | |||||||||
Accounts payable and accrued liabilities | 20,189 | 20,189 | — | — | — | |||||||||
Contingent consideration to ILJIN (4) | 6,332 | 3,582 | 2,289 | 461 | — | |||||||||
Total | 45,622 | 31,101 | 2,780 | 2,602 | 9,139 | |||||||||
(1) | The column identified as due within one year reflects lease inducement payments from the landlord of $406,000 and no rent payable until September 1, 2021 which results in an increase in the lease liability over this period. |
(2) | Operating lease obligations are comprised of the future minimum lease payments for our premises with a lease term of less than one year. |
(3) | We have entered into contractual obligations for services and materials required for our ongoing clinical trials and other R&D projects, our drug supply, and our pre-commercial activities. The purchase obligations presented represent the minimum amount to exit our contractual commitments. |
(4) | Contingent consideration to ILJIN is described in note 7 to the unaudited interim condensed financial statements for the three and nine months ended September 30, 2020. |
We entered into an agreement, effective June 1, 2014, to sublease 5,540 square feet of office and storage space at our head office location in Victoria, British Columbia for a term of five years. On December 6, 2019, the head lessee provided notice to the landlord the intent to terminate the lease effective December 31, 2020.
In March 2020, we entered into a commercial office lease, of 30,531 square feet, for our US commercial center of operations in Rockville, Maryland (MD lease), for an initial term of 11 years. Monthly rent begins in September 2021 and is initially $72,000 per month (escalating to $105,000 over the lease term). As part of the lease agreement, we are entitled to lease inducement payments from the landlord of $2.27 million.
13
During the three months ended September 30, 2020 we entered into an agreement to lease 24,488 square feet of office space for our new corporate headquarter facilities in Victoria, British Columbia. The lease is expected to begin in 2022 for a term of ten years and minimum lease payments for this lease are expected to be $4.31 million. As of September 30, 2020 there has been no accounting recognition associated with this lease, as the Company has not been granted access to the building.
Subsequent to the period ended September 30, 2020 we entered into an agreement to lease premises at #201, 17873 - 106A Avenue, Edmonton, Alberta, consisting of 2,248 square feet of office space, for a term commencing October 1, 2020 to September 30, 2021 at a cost of approximately $2,100 per month.
RELATED PARTY TRANSACTIONS
For the three and nine month periods ended September 30, 2020, Stephen P. Robertson, a partner at Borden Ladner Gervais ("BLG") acts as our corporate secretary. We incurred legal fees in the normal course of business to BLG of $97,000 and $266,000 respectively for the three and nine month periods ended September 30, 2020 compared to $126,000 and $459,000 for the same period in 2019. The amount charged by BLG is based on standard hourly billing rates for the individuals working on our account. For the three and nine month periods ended September 30, 2020, we had no ongoing contractual or other commitments as a result of engaging Mr. Robertson to act as our corporate secretary and Mr. Robertson received no additional compensation for acting as the corporate secretary beyond his standard hourly billing rate. On November 2, 2020 we announced the appointment of Stephen Robertson as our Executive Vice President, General Counsel, Corporate Secretary and Chief Compliance Officer.
The outstanding fair value of contingent consideration payable to ILJIN, an affiliated shareholder and related party, is the result of an Arrangement Agreement (the "ILJIN Agreement") completed on September 20, 2013 between the Company, Aurinia Pharma Corp. and ILJIN.
Pursuant to the terms of the ILJIN Agreement, $10 million in contingent consideration would potentially be owed to ILJIN based on the achievement of future pre-defined clinical and marketing milestones. At September 30, 2020, there is $7.75 million of contingent consideration milestones remaining which we may pay out in the future dependent upon the achievement of the specific pre-defined milestones being met.
The contingent consideration payable to ILJIN is more fully discussed in note 7 of the unaudited interim condensed consolidated financial statements for the three and nine months ended September 30, 2020.
OFF-BALANCE SHEET ARRANGEMENTS
There are no material undisclosed off-balance sheet arrangements that have or are reasonably likely to have, a material current or future effect on our results of operations or financial condition.
CRITICAL ACCOUNTING ESTIMATES AND JUDGMENTS
The preparation of consolidated financial statements in accordance with IFRS often requires management to make estimates about, and apply assumptions or subjective judgment to, future events and other matters that affect the reported amounts of our assets, liabilities, revenues, expenses and related disclosures. Assumptions, estimates and judgments are based on historical experience, expectations, current trends and other factors that management believes to be irrelevant at the time at which our consolidated financial statements are prepared. Management reviews, on a regular basis, our accounting policies, assumptions, estimates and judgments in order to ensure the consolidated financial statements are presented fairly and in accordance with IFRS.
Critical accounting estimates and judgments are those that have a significant risk of causing material adjustment and are often applied to matters or outcomes that are inherently uncertain and subject to change. As such, management cautions that future events often vary from forecasts and expectations and that estimates routinely require adjustment.
In addition the full extent to which the COVID-19 pandemic will directly or indirectly impact the Company’s estimates related to the contingent consideration, lease liability, royalty obligation or results of operations will depend on future developments that are uncertain at this time. As events continue to evolve and additional information becomes available, the Company’s estimates may change materially in future periods.
Management considers the following areas to be those where critical accounting policies affect the significant judgments and estimates used in the preparation of our consolidated financial statements.
Critical estimates in applying Aurinia's accounting policies
• | Contingent consideration |
Contingent consideration is a financial liability recorded at fair value. The amount of contingent consideration to be paid is based on the occurrence of future events, such as the achievement of certain development, regulatory and sales milestones. Accordingly, the estimate of fair value contains uncertainties as it involves judgment about the likelihood and timing of achieving these milestones as
14
well as the discount rate used. Changes in fair value of the contingent consideration obligation result from changes to the assumptions used to estimate the probability of success for each milestone, the anticipated timing of achieving the milestones and the discount period and rate to be applied. A change in any of these assumptions could produce a different fair value, which could have a material impact on the results from operations.
The fair value estimates at September 30, 2020 were based on a weighted average discount rate of 1.6% (December 31, 2019 - 10%) and a presumed payment range between 50% and 90% (December 31, 2019 - 50% and 86%). The decrease of the discount rate was primarily attributable to the significant decline in interest rates caused by the COVID-19 pandemic. The fair value of this contingent consideration as at September 30, 2020 was estimated to be $6.33 million (December 31, 2019 - $5.11 million) and was determined by estimating the probability and timing of achieving the milestones and applying the income approach.
The change in discount rate, increased probability and passage of time, on revaluation, resulted in an increase in contingent consideration of $352,000 and $1.22 million respectively for the three and nine months ended September 30, 2020 compared to an increase in contingent consideration of $127,000 and $207,000 respectively for the three and nine months ended September 30, 2019.
This is a Level 3 recurring fair value measurement. If the probability for success were to increase by a factor of 10% for each milestone, this would increase the net present value (NPV) of the obligation by approximately $760,000 as at September 30, 2020. If the probability for success were to decrease by a factor of 10% for each milestone, this would decrease the NPV of the obligation by approximately $762,000 as at September 30, 2020. If the discount rate were to increase by 2%, this would decrease the NPV of the obligation by approximately $134,000. If the discount rate were to decrease by 2%, this would increase the NPV of the obligation by approximately $140,000.
• | Royalty obligation |
As the royalty obligation is a calculation of future payments the Company is required to use judgment to determine the most appropriate model to use to measure the obligation and is required to use significant judgment and estimates in determining the inputs into the model. There are multiple unobservable inputs. The determination of these cash flows is subject to significant estimates and assumptions including:
• | Net pricing - this includes estimates of the gross pricing of the product, gross to net discounts and annual price escalations of the product |
• | Number of patients being treated - this includes various inputs to derive the number of patients receiving treatment including the number of patients receiving treatment, market penetration, time to peak market penetration, and the timing of generics entering the market |
• | Probability of success and occurrence - this is the probability of the future cash outflows occurring |
• | Discount rate - the rate selected to measure the risks inherent in the future cash flows |
Management developed the model and inputs in conjunction with the internal scientific team and utilized third party scientific studies, information provided by third party consultants engaged by the Company and research papers as sources to develop the inputs. They also utilized the market capitalization of the Company as one input into the model. Management believes the liability is based on reasonable assumptions, however, these assumptions may be incomplete or inaccurate and unanticipated events and circumstances may occur. Reasonable possible changes in the assumptions may have a material impact on the estimated value of the obligation. There are numerous significant inputs into the model all of which individually or in combination could result in a material change to the obligation.
As a result of the completion and results obtained of the AURORA trial in the fourth quarter of 2019 we re-assessed the probability of royalty obligation payments being required in the future, and have recorded the royalty obligation at December 31, 2019. Until one of the triggering events occur, no royalty payments are required to be paid. No material royalties on sales or licensing are expected to be paid in the next twelve months and therefore the royalty obligation has been classified as long term. The fair value of the royalty obligation as at September 30, 2020 was estimated to be $8 million (December 31, 2019 - $7.20 million).
We are required to use significant judgment and estimates in determining the inputs into the model. The key assumptions used by management include the estimated probability of market approval of 90%, and the discount rate of 11%. If the probability of success were to increase to 99% this would increase the obligation by $799,000 and if it were to decrease to 81% this would decrease the obligation by $799,000. If the discount rate were to increase to 12.1%, this would decrease the obligation by $559,000, and if it were to decrease to 9.9%, this would increase the obligation by $615,000. An increase in the estimated gross pricing or number of patients being treated by 10% would result in an increase in the obligation of $793,000 while a decrease in the estimated gross pricing or number of patients being treated by 10% would result in a decrease in the obligation of $793,000.
15
• | Derivative warrant liabilities |
Warrants issued pursuant to equity offerings that are potentially exercisable in cash or on a cashless basis resulting in a variable number of shares being issued are considered derivative liabilities and therefore measured at fair value.
We use the Black-Scholes pricing model to estimate fair value at each exercise and period end date. The key assumptions used in the model are the expected future volatility in the price of our shares and the expected life of the warrants. The impact of changes in key assumptions are noted below.
These derivative warrant liabilities are Level 3 recurring fair value measurements.
The key Level 3 inputs used by management to estimate the fair value are the market price and the expected volatility. If the market price were to increase by a factor of 10%, this would increase the estimated fair value of the obligation by approximately $2.48 million as at September 30, 2020. If the market price were to decrease by a factor of 10%, this would decrease the estimated fair value of the obligation by approximately $2.48 million. If the volatility were to increase by 10%, this would increase the estimated fair value of the obligation by approximately $14,000. If the volatility were to decrease by 10%, this would decrease estimated fair value of the obligation by approximately $6,000 as at September 30, 2020.
• | Fair value of stock options |
Determining the fair value of stock options on the grant date requires judgment related to the choice of a pricing model, the estimation of stock price volatility and the expected term of the underlying instruments. Any changes in the estimates or inputs utilized to determine fair value could result in a significant impact on our reported operating results, liabilities or other components of shareholders’ equity. The key assumptions used by management is the stock price volatility.
If the stock price volatility was higher by a factor of 10% on the option grant dates in 2020, this would have increased annual stock compensation expense by approximately $278,000. If the stock price volatility was lower by a factor of 10% on the grant date, this would have decreased annual stock compensation expense by approximately $281,000.
We use the Black-Scholes option pricing model to estimate the fair value of the options granted. We consider historical volatility of our Common Shares in estimating its future stock price volatility. The risk-free interest rate for the expected life of the options was based on the yield available on government benchmark bonds with an approximate equivalent remaining term at the time of the grant. The expected life is based upon expected employee exercise and expected post-vesting employment termination behavior.
Critical judgments in applying Aurinia's accounting policies
• | Revenue recognition |
Our assessments related to the recognition of revenues for arrangements containing multiple elements are based on estimates and assumptions. Judgment is necessary to identify separate performance obligations and to allocate related consideration to each separate performance obligation. Where deferral of license fees is deemed appropriate, subsequent revenue recognition is often determined based on certain assumptions and estimates, our continuing involvement in the arrangement, the benefits expected to be derived by the customer and expected patent lives. The estimate of variable consideration requires significant judgment and an assessment of their potential reversal. We also use judgment in assessing if a license is a right to use or a right to access intellectual property. Factors that are considered include whether the customer reasonably expects (arising from the entity's customary business practices) that the entity will undertake activities that will significantly affect the intellectual property, the rights granted by the license directly expose the customer to any positive or negative effects of the entity's activities and whether those activities transfer a separate good or service to the customer. To the extent that any of the key assumptions or estimates change, future operating results could be affected.
•Lease liability - Determining the lease term
In determining the lease term, management considers all facts and circumstances that create an economic incentive to exercise an extension option, or not exercise a termination option. Extension options (or periods after termination options) are only included in the lease term if the lease is reasonably certain to be extended (or not terminated).
For leases of office space, the following factors are the most relevant:
• | If there are significant penalties to terminate (or not extend), the Company is typically reasonably certain to extend (or not terminate). |
• | If any leasehold improvements are expected to have significant remaining value, the Company is typically reasonably certain to extend (or not terminate). |
Otherwise, we consider other factors including historical lease durations, government incentives received in connection with the lease, and business disruption required to replace the leased asset or relocate facilities. Most extension options in office leases have not been included in the lease liability, because we could replace the leasehold improvement assets and relocate facilities without significant cost or business disruption.
16
•Royalty obligation
The Company follows the guidance of IAS 19 in assessing the recognition of a royalty obligation. The recognition of a royalty obligation and the determination of the amount to record is based on estimates and assumptions. Judgment is necessary to determine these estimates and assumptions which include determining the likelihood of future material payments becoming probable and the best methods by which to quantify these payments.
On December 4, 2019 we released positive AURORA Phase 3 trial results for LN and on July 21, 2020 we announced that the FDA has accepted the filing of the NDA for voclosporin, as a potential treatment for LN. As a result, management has determined that royalties are more probable to be payable in the future than in previous years, and therefore have recorded a royalty obligation.
Management determined that an income approach using an internal risk-adjusted net present value analysis was the best estimate to measure the royalty obligation. This approach was further supported by a valuation model utilizing a market capitalization approach.
• | Impairment of intangible assets |
We follow the guidance of IAS 36 to determine when impairment indicators exist for intangible assets. When impairment indicators exist, we are required to make a formal estimate of the recoverable amount of its intangible assets. This determination requires significant judgment. In making this judgment, management evaluates external and internal factors, such as significant adverse changes in the technological, market, economic or legal environment in which we operate as well as the results of our ongoing development programs. Management also considers the carrying amount of our net assets in relation to our market capitalization as a key indicator. In making a judgment as to whether impairment indicators exist as at September 30, 2020, management concluded there were none.
• | Derivative warrant liabilities |
Management has determined that derivative warrant liabilities are classified as long term as these derivative warrant liabilities will ultimately be settled for Common Shares and therefore the classification is not relevant.
• | Capitalization of R&D expense and inventory |
Internal development expenditure is capitalized if it meets the recognition criteria of IAS 38 Intangible Assets. This is considered a key judgment.
Judgment is applied in determining the starting point for capitalizing internal development costs. However, a strong indication that the criteria in IAS 38 to capitalize these costs arises when a product is likely to obtain final approval by a regulatory authority. It is the clearest point at which the technical feasibility of completing the asset is proven and is the most difficult criterion to demonstrate. Filing for obtaining regulatory approval is also sometimes considered as the point at which all relevant criteria including technical feasibility are considered met. During 2019 the Company successfully completed the phase 3 trial for lupus nephritis. As of September 30, 2020, the Company had completed an NDA with the FDA for voclosporin as a potential treatment for LN, but has not received regulatory approval in any market. However, management has determined that regulatory approval of voclosporin, as a potential treatment for LN is reasonably assured; therefore, in management's judgment the criteria to capitalize development costs has been met.
• | Deferred tax asset |
The Company recognizes deferred tax assets only to the extent that it is probable that future taxable profits, feasible tax planning strategies and deferred tax liabilities will be available against which the tax losses can be utilized. Estimation of the level of future taxable profits is therefore required in order to determine the appropriate carrying value of the deferred tax asset. Given the Company's past losses, plans to continue R&D in other indications and uncertainty of its ability to generate future taxable profit, management does not believe that it is more probable than not that the Company can realize its deferred tax assets and therefore, it has not recognized any amount in the consolidated statements of financial position.
RISKS AND UNCERTAINTIES
We have invested a significant portion of our time and financial resources in the development of voclosporin. We anticipate that our ability to generate revenues and meet expectations will depend primarily on the successful development, regulatory approval and commercialization of voclosporin.
The successful development and commercialization of voclosporin will depend on several factors, including the following:
• | receipt of marketing approvals from the FDA and other regulatory authorities with a commercially viable label; |
• | securing and maintaining sufficient expertise and resources to help in the continuing development and eventual commercialization of voclosporin; |
• | maintaining suitable manufacturing and supply arrangements to ensure commercial quantities of the product through validated processes; |
• | acceptance and adoption of the product by the medical community and third-party payers; and |
• | our ability to raise future financial resources when required. |
17
A more detailed list of the risks and uncertainties affecting us can be found under the heading "Risk Factors" in our annual information form which is filed on SEDAR and EDGAR. There have been no material changes from the risk factors set forth in our annual information form other than:
COVID-19
In December 2019, a novel strain of coronavirus, COVID-19, surfaced in Wuhan, China. On January 30, 2020, the World Health Organization declared the COVID-19 outbreak a "Public Health Emergency of International Concern" and on March 11, 2020, declared it to be a pandemic. Actions taken around the world to help mitigate the spread of the coronavirus include restrictions on travel, and quarantines in certain areas, and forced closures for certain types of public places and businesses. Public health officials have recommended and mandated precautions to mitigate the spread of COVID-19, including prohibitions on non-essential travel, congregating in heavily populated areas and shelter-in-place orders or similar measures. In response to the spread of COVID-19 we have implemented a remote working environment in order to comply with these public health actions.
It is unknown how long the adverse conditions associated with the coronavirus will last and what the complete financial effect will be to the Company. Due to the actions taken by the public health officials, we may face delays and difficulties enrolling or retaining patients in our clinical trials if patients are affected by the virus or are unable to travel to our clinical trial sites.
As a result of the COVID-19 pandemic, we may experience disruptions that could result in:
• | delays or difficulties in enrolling patients in our clinical trials; |
• | delays or difficulties in building out commercial infrastructure; |
• | delays in recruiting for key positions; |
• | delays or difficulties in clinical site initiation, including difficulties in recruiting clinical site investigators and clinical site staff; |
• | interruption of key clinical trial activities, such as clinical trial site data monitoring, due to limitations on travel imposed or recommended by federal, provincial or state governments, employers and others or interruption of clinical trial subject visits and study procedures, which may impact the integrity of subject data and clinical study endpoints; |
• | interruption or delays in the operations of the FDA, which may impact approval timelines; |
• | interruption of, or delays in receiving supplies of our product candidates from our contract manufacturing organizations due to staffing shortages, production slowdowns or stoppages and disruptions in delivery systems; and |
• | limitations on employee resources that would otherwise be focused on the conduct of our preclinical studies and clinical trials, including because of sickness of employees or their families or the desire of employees to avoid contact with large groups of people. |
To the extent the COVID-19 pandemic adversely affects our business and financial results, it may also have the effect of heightening many of the other risks described in this MD&A and in the "Risk Factors" section of our Annual Information Form. Because of the highly uncertain and dynamic nature of events relating to the COVID-19 pandemic, it is not currently possible to estimate the impact of COVID-19 on our business. However, these effects could have a material impact on our operations, and we will continue to monitor the COVID-19 situation.
The results of pre-clinical studies and initial clinical trials are not necessarily predictive of future results, and our current product candidate may not have favourable results in later trials or in the commercial setting.
Success in pre-clinical or animal studies and early clinical trials neither ensure that later large-scale efficacy trials will be successful, nor does it predict final results. Pre-clinical tests and Phase 1 and Phase 2 clinical trials are primarily designed to test safety, to study pharmacokinetics and pharmacodynamics and to understand the side effects of product candidates at various doses and schedules. Favourable results in early trials may not be repeated in later trials.
A number of companies in the life sciences industry have suffered significant setbacks in advanced clinical trials, even after positive results in earlier trials. Clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals. Negative or inconclusive results or adverse medical events during a clinical trial could cause a clinical trial to be delayed, repeated or terminated. In addition, failure to construct appropriate clinical trial protocols could result in the test or control group experiencing a disproportionate number of adverse events and could cause a clinical trial to be repeated or terminated.
Pre-clinical data and the clinical results we have obtained for voclosporin (for LN or any other indication) may not predict results from studies in larger numbers of subjects drawn from more diverse populations or in a commercial setting, and also may not predict the ability of our product to achieve its intended goals, or to do so safely.
We will be required to demonstrate in Phase 3 clinical trials that voclosporin is safe and effective for use in a diverse population before we can seek regulatory approvals for its commercial sale. There is typically an extremely high rate of attrition from the failure of product candidates proceeding through clinical and post approval trials. If voclosporin fails to demonstrate sufficient safety and efficacy in ongoing or future clinical trials, we could experience potentially significant delays in, or be required to abandon development of, our product candidate currently under development.
18
We could be subject to litigation, allegations or other legal claims.
Our assets or our business activities may be subject to disputes that may result in litigation or other legal claims. We may be subject to allegations through press, social media, the courts or other mediums that may or may not be founded. We may be required to respond to or defend against these claims and/or allegations, which will divert resources away from our principal business. There can be no assurance that our defense of such claims and/or allegations would be successful, and we may be required to make material settlements. This could have a material adverse effect on our business prospects, results of operations, cash flows, financial condition and corporate reputation.
Capital management
Our objective in managing capital, consisting of shareholders' equity, with cash, cash equivalents and short term and long term investments being its primary components, is to ensure sufficient liquidity to fund pre-commercialization and launch activities, R&D activities, corporate, administration and business development expenses and working capital requirements. This objective has remained the same from that of the previous year.
Over the past two years, we have raised capital via public offerings, the exercise of warrants and stock options and draw-downs under our ATM facilities, as our primary sources of liquidity.
As our policy is to retain cash to keep funds available to finance the activities required to advance our product development and to fund pre-commercialization and launch activities of voclosporin as a treatment for LN, we do not currently pay dividends.
We are not subject to any capital requirements imposed by any regulators or by any other external source.
Financial instruments and Risks
We are exposed to credit risks and market risks related to changes in interest rates and foreign currency exchange, each of which could affect the value of our current assets and liabilities.
We have invested our cash reserves in U.S. dollar denominated, fixed rate, highly liquid and highly rated financial instruments such as treasury notes, banker acceptances, corporate bonds, and term deposits. As a result of the COVID-19 pandemic, interest rates have significantly decreased and while we do not believe the results of operations or cash flows will be significantly impacted by this interest rate decrease, interest earned on our cash balances is expected to decrease from previous levels. In addition, we have adjusted the interest rates used in determining the fair valuation of the contingent consideration and royalty obligation at September 30, 2020.
Financial risk factors
Our activities can expose us to a variety of financial risks: market risk (including currency risk and interest rate risk), credit risk and liquidity risk. Risk management is carried out by management under policies approved by the Board of Directors. Management identifies and evaluates the financial risks. Our overall risk management program seeks to minimize adverse effects on our financial performance.
Liquidity risk
Liquidity risk is the risk we will not be able to meet our financial obligations as they fall due. We manage our liquidity risk through the management of our capital structure and financial leverage, as discussed above in "Capital Management". We also manage liquidity risk by continuously monitoring actual and projected cash flows. The Board of Directors reviews and approves our budget, as well as any material transactions out of the ordinary course of business. We invest our cash equivalents in U.S. denominated term deposits with 30 to 90-day maturities, and U.S. denominated short term and long term investments consisting of bonds and treasury notes issued by banks and/or United States or Canadian governments with maturities not exceeding two years to ensure our liquidity needs are met.
All of our financial liabilities are due within one year except for the long-term portion of the contingent consideration, as described in note 7 to the unaudited interim condensed consolidated financial statements for the three and nine months ended September 30, 2020, the lease liability as described in note 8 to the unaudited interim condensed consolidated financial statements for the three and nine months ended September 30, 2020, the royalty obligation, in note 9 to the unaudited interim condensed consolidated financial statements for the three and nine months ended September 30, 2020, and the derivative warrant liability, note 10 to the unaudited interim condensed consolidated financial statements for the three and nine months ended September 30, 2020.
Interest rate risk
Interest rate risk is the risk the fair value or future cash flows of a financial instrument will fluctuate because of changes in market interest rates.
Financial assets and financial liabilities with variable interest rates expose us to cash flow interest rate risk. Our cash and cash equivalents are comprised of highly liquid investments that earn interest at market rates and the short term investments held during the year were comprised of bank or government bonds with a maturity of two years or less. Accounts receivable, accounts payable and accrued liabilities bear no interest.
We manage our interest rate risk by maintaining the liquidity necessary to conduct operations on a day-to-day basis.
19
Credit risk
Financial instruments that potentially subject us to significant concentrations of credit risk consist principally of cash, cash equivalents and short term and long term investments which are held at major Canadian and US banks. We regularly monitor the credit risk exposure and take steps to mitigate the likelihood of these exposures resulting in expected loss.
Foreign currency risk
We are exposed to financial risk related to the fluctuation of foreign currency exchange rates. Foreign currency risk is the risk variations in exchange rates between the US dollars and foreign currencies, primarily with the Canadian dollar, will affect our operating and financial results.
The following table presents our exposure to the Canadian dollar:
(in thousands) | |||||
September 30, 2020 $ | September 30, 2019 $ | ||||
Cash | 12,733 | 155 | |||
Accounts receivable | 25 | 89 | |||
Accounts payable and accrued liabilities | (2,473 | ) | (1,669 | ) | |
Net exposure | 10,285 | (1,425 | ) | ||
Reporting date rate | |||||
September 30, 2020 $ | September 30, 2019 $ | ||||
CA$ – US$ | 0.747 | 0.755 | |||
Our cash held in Canadian dollars was the result of the exercise of stock options which were denominated in Canadian dollars.
As we require Canadian dollars for our ongoing Canadian operational expenses, we maintain a small percentage of our overall cash and cash equivalents in Canadian dollars.
Based on our foreign currency exposure noted above, varying the foreign exchange rates to reflect a 10% strengthening of the CA$ would have increased the net loss by $770,000 assuming all other variables remained constant. An assumed 10% weakening of the CA$ would have had an equal but opposite effect to the amounts shown above, on the basis all other variables remain constant.
Intellectual Property Rights
Patents and other proprietary rights are essential to our business. Our policy has been to file patent applications to protect technology, inventions and improvements to our inventions that are considered important to the development of our business.
We have an extensive granted patent portfolio covering voclosporin, including granted United States patents, for composition of matter, methods of use, formulations and synthesis. The corresponding Canadian, South African and Israeli patents are owned by Paladin Labs Inc. We anticipate that upon regulatory approval, patent protection for voclosporin will be extended in the United States (Patent Term Extension) and certain other major markets, including Europe and Japan, until at least October 2027 under the Hatch-Waxman Act in the United States and comparable patent extension laws in other countries (including the Supplementary Protection Certificate program in Europe). Opportunities may also be available to add an additional six months of exclusivity related to pediatric studies which are currently in the planning process. In addition to patent rights, we also expect to receive “new chemical entity” exclusivity for voclosporin in certain countries, which provides this type of exclusivity for five years in the United States and up to ten years in Europe.
Further, on May 14, 2019 Aurinia was granted U.S. Patent No. 10,286,036 with a term extending to December 2037, with claims directed at our voclosporin dosing protocol for LN. The allowed claims broadly cover the novel voclosporin individualized flat-dosed pharmacodynamic treatment protocol adhered to and required in both the previously reported Phase 2 AURA-LV trial and our Phase 3 confirmatory AURORA clinical trial. Notably, the allowed claims cover a method of modifying the dose of voclosporin in patients with LN based on patient specific pharmacodynamic parameters. If the FDA approves the use of voclosporin for LN and the label for such use follows the dosing protocol claimed in U.S. Patent No. 10,286,036, this patent will expand the scope of intellectual property protection for voclosporin, which already includes manufacturing, formulation, synthesis and composition of matter patents. We have also filed for protection of this subject matter under the PCT and have the option of applying for similar protection in the member countries thereof. This may lead to the granting of similar claims in major global pharmaceutical markets.
We have licensed the development and distribution rights to voclosporin for China, Hong Kong and Taiwan to 3SBio. This license is royalty bearing and we will also supply finished product to 3SBio on a cost-plus basis. We do not expect to receive any royalty revenue pursuant to this license in the foreseeable future.
20
We have invested a significant portion of our time and financial resources in the development of voclosporin. We anticipate that our ability to generate revenues and meet expectations will depend primarily on the successful development, regulatory approval and commercialization of voclosporin.
CONTINGENCIES
We may, from time to time, be subject to claims and legal proceedings brought against us in the normal course of business. Such matters are subject to many uncertainties. Management believes that the ultimate resolution of such contingencies will not have a material adverse effect on our consolidated financial position.
We have entered into indemnification agreements with our officers and directors. The maximum potential amount of future payments required under these indemnification agreements is unlimited. However, we do maintain liability insurance to limit our exposure.
We have entered into license and R&D agreements with third parties that include indemnification and obligation provisions that are customary in the industry. These guarantees generally require us to compensate the other party for certain damages and costs incurred as a result of third party claims or damages arising from these transactions. These provisions may survive termination of the underlying agreement. The nature of the obligations prevents us from making a reasonable estimate of the maximum potential amount we could be required to pay. Historically, we have not made any payments under such agreements and no amount has been accrued in the accompanying audited consolidated financial statements.
DISCLOSURE CONTROLS AND PROCEDURES AND INTERNAL CONTROL OVER FINANCIAL REPORTING
Aurinia's management is responsible for establishing and maintaining adequate internal control over financial reporting ("ICFR") and disclosure controls and procedures ("DC&P").
ICFR is a framework designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with IFRS. Management has used the Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) in order to assess the effectiveness of the Company's ICFR.
DC&P form a broader framework designed to provide reasonable assurance the information required to be disclosed by Aurinia in its annual and interim filings and other reports filed under securities legislation is recorded, processed, summarized and reported within the time frame specified in securities legislation and includes controls and procedures designed to ensure that information required to be disclosed by Aurinia in its annual and interim filings and other reports submitted under securities legislation is accumulated and communicated to our management to allow timely decisions regarding required disclosure.
Together, the ICFR and DC&P frameworks provide internal control over financial reporting and disclosure. We maintain disclosure controls and procedures that are designed to provide reasonable assurance that information, which is required to be disclosed in our annual and interim filings and other reports filed under securities legislation, is accumulated and communicated in a timely fashion. Due to their inherent limitations, Aurinia acknowledges that, no matter how well designed, ICFR and DC&P can provide only reasonable assurance of achieving the desired control objectives and as such may not prevent or detect all misstatements. Further, the effectiveness of ICFR is subject to the risk that controls may become inadequate because of changes in conditions or that the degree of compliance with policies or procedures may change.
There have been no significant changes to our disclosure controls nor to our internal controls over financial reporting for the three and nine month periods ended September 30, 2020 that have materially affected, or are reasonably likely to materially affect, the reliability of financial reporting.
21
UPDATED SHARE INFORMATION
As at November 5, 2020, the following class of shares and equity securities potentially convertible into Common Shares were outstanding:
(in thousands) | ||
Common Shares | 126,568 | |
Convertible equity securities | ||
Derivative liability warrants | 1,690 | |
Stock options | 11,443 | |
Performance awards | 439 | |
Subsequent to September 30, 2020, we issued 118,000 Common Shares upon the exercise of 118,000 stock options for proceeds of $463,000. We also issued 118,000 stock options to new employees at a weighted average exercise price of $14.73 (CA $19.58). Of the subsequently issued stock options, 96,000 were inducement stock options to nine new employees entering into employment with the Company in accordance with Nasdaq listing rule 5635(c)(4) at a price of $14.73 (CA$19.57). For the purpose of TSX approval, the Company relied on the exemption set forth in Section 602.1 of the TSX Company Manual, which provides that the TSX will not apply its standards to certain transactions involving eligible inter-listed issuers on a recognized exchange. These options are recorded outside of the stock option plan. We also granted 439,000 performance awards (PAs) to officers of the Company, which will vest as the predefined performance milestones are met.
SUPPLEMENTAL INFORMATION
Quarterly Information
(expressed in thousands except per share data)
Set forth below is selected unaudited consolidated financial data for each of the last eight quarters:
2020 | 2019 | 2018 | ||||||||||||||
Sept 30 | Jun 30 | Mar 31 | Dec 31 | Sept 30 | Jun 30 | Mar 31 | Dec 31 | |||||||||
Revenues | 29 | 29 | 30 | 29 | 230 | 29 | 30 | 29 | ||||||||
Expenses | ||||||||||||||||
R&D | 4,800 | 11,076 | 13,835 | 13,292 | 17,791 | 11,152 | 10,631 | 10,839 | ||||||||
Corporate, administration and business development | 31,068 | 15,541 | 11,061 | 7,246 | 6,061 | 4,946 | 3,901 | 3,498 | ||||||||
Amortization of tangible and intangible assets | 502 | 493 | 403 | 391 | 389 | 385 | 383 | 355 | ||||||||
Other expenses | 426 | (287 | ) | 2,212 | 7,963 | 140 | 833 | 55 | (65 | ) | ||||||
Total expenses | 36,796 | 26,823 | 27,511 | 28,892 | 24,381 | 17,316 | 14,970 | 14,627 | ||||||||
Loss before interest income , finance costs, change in estimated fair value of derivative warrant liabilities and income taxes | (36,767 | ) | (26,794 | ) | (27,481 | ) | (28,863 | ) | (24,151 | ) | (17,287 | ) | (14,940 | ) | (14,598 | ) |
Interest income | 170 | 320 | 891 | 479 | 636 | 787 | 800 | 671 | ||||||||
Finance costs | (101 | ) | (78 | ) | (25 | ) | (9 | ) | (9 | ) | (10 | ) | (11 | ) | — | |
Net loss before change in estimated fair value of derivative warrant liabilities and income taxes | (36,698 | ) | (26,552 | ) | (26,615 | ) | (28,393 | ) | (23,524 | ) | (16,510 | ) | (14,151 | ) | (13,927 | ) |
Change in estimated fair value of derivative warrant liabilities | 2,599 | (2,952 | ) | 9,845 | (47,986 | ) | 4,512 | 625 | 1,725 | (593 | ) | |||||
Income tax recovery (expense) | 35 | (22 | ) | 236 | (90 | ) | (25 | ) | (16 | ) | (13 | ) | (73 | ) | ||
Net loss for the period | (34,064 | ) | (29,526 | ) | (16,534 | ) | (76,469 | ) | (19,037 | ) | (15,901 | ) | (12,439 | ) | (14,593 | ) |
Net loss per Common Share – basic and diluted | (0.28 | ) | (0.26 | ) | (0.15 | ) | (0.78 | ) | (0.21 | ) | (0.17 | ) | (0.14 | ) | (0.17 | ) |
Common Shares outstanding | 126,450 | 112,705 | 112,487 | 111,798 | 94,285 | 91,793 | 91,646 | 85,500 | ||||||||
Weighted average number of Common Shares outstanding | 122,357 | 112,576 | 112,209 | 97,936 | 92,169 | 91,768 | 90,146 | 85,384 | ||||||||
Previously, interest income and finance costs were labeled on the statement of operations and comprehensive loss as other expenses. In 2020, 2019 and 2018 they have been disaggregated and relabeled as interest income and finance costs in the table above.
22
Summary of Quarterly Results
The primary factors affecting the magnitude of our losses in the various quarters are noted below and include the timing of R&D costs associated with the clinical development program, timing and amount of stock compensation expense, and fluctuations in the non-cash change in estimated fair value of derivative warrant liabilities.
The decrease in the R&D expense for the three months ended September 30, 2020 was primarily due to the reduced R&D activities, including CRO costs following the completion of the AURORA trial during the period ended June 30, 2020 in conjunction with the capitalization of development costs, inventory and the reversal of the inventory provision related to previously expensed drug substance manufacturing costs. The increase in the R&D expense for the three months ended September 30, 2019 primarily reflected the cost of manufacturing active drug substance batches which will be used for future commercial use upon marketing approval.
The increase in corporate, administration and business development expenses for the three months ended September 30, 2020 and the three months ended June 30, 2020 are due to the development of our commercial capabilities across the organization including the expansion of the commercial team and higher professional and consulting fees related to recruiting, legal, tax advice, human resources, information technology and pre-commercialization activities including market research, market access, patience advocacy and communications. The increase reflected significant expansion in activity levels across the organization.
Corporate, administration and business development expenses also included non-cash stock-based compensation expense of $3.75 million for the three months ended September 30, 2020, non-cash stock-based compensation expense of $3.12 million for the three months ended June 30, 2020, non-cash stock-based compensation expense of $2.28 million for the three months ended March 31, 2020, non-cash stock-based compensation expense of $1.34 million for the three months ended December 31, 2019, non-cash stock-based compensation expense of $1.43 million for the three months ended September 30, 2019 compared to $1.21 million for the three months ended June 30, 2019, $742,000 for the three months ended March 31, 2019, $686,000 for the three months ended December 31, 2018, and $887,000 for the three months ended September 30, 2018.
Other expenses for the three months ended September 30, 2020 included a $352,000 adjustment on contingent consideration, $300,000 adjustment for the royalty obligation and a $255,000 foreign exchange gain. Other expenses for the three months ended June 30, 2020 included a $271,000 adjustment on contingent consideration, $100,000 adjustment for the royalty obligation and a $458,000 foreign exchange gain. Other expenses for the three months ended March 31, 2020 included royalty obligation expense of $600,000, a $596,000 revaluation adjustment on contingent consideration and a foreign exchange loss of $1.02 million.
Other expenses for the three months ended December 31, 2019 included royalty obligation expense of $7.20 million as discussed in the "Results of operations-other expenses" section of this MD&A and a $978,000 revaluation adjustment on contingent consideration.
Other expenses for the three months ended June 30, 2019 included $720,000 of costs associated with the successful defense of a proxy contest for our June 26, 2019 annual general meeting.
We record non-cash adjustments each quarter resulting from the fair value revaluation of the derivative warrant liabilities. These revaluations fluctuate based primarily on the market price of our Common Shares. An increase in the market price of our Common Shares results in a loss on revaluation while a decrease results in a gain on revaluation.
The change in the estimated fair value of the derivative warrant liabilities for the three months ended September 30, 2020 of $2.60 million reflected a decrease in our share price to $14.73 per Common Share at September 30, 2020 compared to $16.25 per Common Share at June 30, 2020. The change in the estimated fair value of the derivative warrant liabilities for the three months ended June 30, 2020 of $2.95 million reflected an increase in our share price to $16.25 per Common Share at June 30, 2020 compared to $14.51 per Common Share at March 31, 2020. The change in the estimated fair value of the derivative warrant liabilities for the three months ended March 31, 2020 of $9.85 million reflected a decrease in our share price to $14.51 per Common Share at March 31, 2020 compared to $20.26 per Common Share at December 31, 2019. The change in the estimated fair value of the derivative warrant liabilities for the three months ended December 31, 2019 of $47.99 million reflected an large increase in our share price to $20.26 per Common Share at December 31, 2019 and an increased share price when 1.83 million warrants were exercised in December 2019, compared to $5.34 per Common Share at September 30, 2019. The change in the estimated fair value of the derivative warrant liabilities for the three months ended September 30, 2019 of $4.51 million reflected a decrease in our share price to $5.34 per Common Share at September 30, 2019 compared to $6.58 per Common Share at June 30, 2019 and a reduction in the annualized volatility to 33% at September 30, 2019 from 40% at June 30, 2019.
Aurinia Pharmaceuticals Inc.
1203 - 4464 Markham Street
Victoria, BC V8Z 7X8
www.auriniapharma.com
23